Jump to
S1C2
S1C3
S1C4
Energy calculated at wB97X-D/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -580.030334 |
| Energy at 298.15K | |
| HF Energy | -580.030334 |
| Nuclear repulsion energy | 67.756505 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σg |
2364 |
2258 |
0.00 |
276.97 |
0.30 |
0.46 |
| 2 |
Σg |
792 |
757 |
0.00 |
118.02 |
0.18 |
0.31 |
| 3 |
Σu |
2359 |
2254 |
0.02 |
0.00 |
0.00 |
0.00 |
| 4 |
Πg |
547i |
523i |
0.00 |
25.68 |
0.75 |
0.86 |
| 4 |
Πg |
547i |
523i |
0.00 |
25.68 |
0.75 |
0.86 |
| 5 |
Πu |
468 |
447 |
1.52 |
0.00 |
0.00 |
0.00 |
| 5 |
Πu |
468 |
447 |
1.52 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 2678.9 cm
-1
Scaled (by 0.9552) Zero Point Vibrational Energy (zpe) 2558.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/Def2TZVPP
Point Group is D∞h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
0.000 |
0.976 |
| Si2 |
0.000 |
0.000 |
-0.976 |
| H3 |
0.000 |
0.000 |
2.435 |
| H4 |
0.000 |
0.000 |
-2.435 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 1.9516 | 1.4588 | 3.4104 |
Si2 | 1.9516 | | 3.4104 | 1.4588 | H3 | 1.4588 | 3.4104 | | 4.8692 | H4 | 3.4104 | 1.4588 | 4.8692 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
180.000 |
|
Si2 |
Si1 |
H3 |
180.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
-0.080 |
|
|
|
| 2 |
Si |
-0.080 |
|
|
|
| 3 |
H |
0.080 |
|
|
|
| 4 |
H |
0.080 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.032 |
0.000 |
0.000 |
| y |
0.000 |
-30.032 |
0.000 |
| z |
0.000 |
0.000 |
-19.831 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-5.100 |
0.000 |
0.000 |
| y |
0.000 |
-5.100 |
0.000 |
| z |
0.000 |
0.000 |
10.200 |
|
| Polar |
|---|
| 3z2-r2 | 20.401 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.637 |
0.000 |
0.000 |
| y |
0.000 |
6.637 |
0.000 |
| z |
0.000 |
0.000 |
14.834 |
<r2> (average value of r
2) Å
2
| <r2> |
55.149 |
| (<r2>)1/2 |
7.426 |
Jump to
S1C1
S1C3
S1C4
Energy calculated at wB97X-D/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -580.056963 |
| Energy at 298.15K | |
| HF Energy | -580.056963 |
| Nuclear repulsion energy | 64.747996 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2188 |
2090 |
0.00 |
385.05 |
0.43 |
0.60 |
| 2 |
Ag |
631 |
603 |
0.00 |
55.37 |
0.45 |
0.62 |
| 3 |
Ag |
564 |
539 |
0.00 |
71.19 |
0.02 |
0.04 |
| 4 |
Au |
183 |
175 |
71.05 |
0.00 |
0.00 |
0.00 |
| 5 |
Bu |
2192 |
2094 |
117.93 |
0.00 |
0.00 |
0.00 |
| 6 |
Bu |
181 |
173 |
71.63 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 2969.8 cm
-1
Scaled (by 0.9552) Zero Point Vibrational Energy (zpe) 2836.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/Def2TZVPP
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.037 |
0.000 |
| Si2 |
0.000 |
-1.037 |
0.000 |
| H3 |
1.200 |
1.916 |
0.000 |
| H4 |
-1.200 |
-1.916 |
0.000 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 2.0734 | 1.4878 | 3.1874 |
Si2 | 2.0734 | | 3.1874 | 1.4878 | H3 | 1.4878 | 3.1874 | | 4.5218 | H4 | 3.1874 | 1.4878 | 4.5218 | |
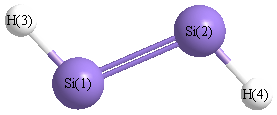 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
126.232 |
|
Si2 |
Si1 |
H3 |
126.232 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
-0.007 |
|
|
|
| 2 |
Si |
-0.007 |
|
|
|
| 3 |
H |
0.007 |
|
|
|
| 4 |
H |
0.007 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-28.837 |
-0.161 |
0.000 |
| y |
-0.161 |
-22.210 |
0.000 |
| z |
0.000 |
0.000 |
-30.671 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.396 |
-0.161 |
0.000 |
| y |
-0.161 |
7.544 |
0.000 |
| z |
0.000 |
0.000 |
-5.148 |
|
| Polar |
|---|
| 3z2-r2 | -10.296 |
|---|
| x2-y2 | -6.627 |
|---|
| xy | -0.161 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.845 |
0.089 |
0.000 |
| y |
0.089 |
15.885 |
0.000 |
| z |
0.000 |
0.000 |
7.827 |
<r2> (average value of r
2) Å
2
| <r2> |
57.330 |
| (<r2>)1/2 |
7.572 |
Jump to
S1C1
S1C2
S1C4
Energy calculated at wB97X-D/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -580.097040 |
| Energy at 298.15K | |
| HF Energy | -580.097040 |
| Nuclear repulsion energy | 65.388158 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
1642 |
1568 |
2.01 |
89.11 |
0.03 |
0.06 |
| 2 |
A1 |
955 |
913 |
27.92 |
11.66 |
0.26 |
0.42 |
| 3 |
A1 |
568 |
542 |
0.21 |
65.22 |
0.29 |
0.45 |
| 4 |
A2 |
1111 |
1061 |
0.00 |
6.22 |
0.75 |
0.86 |
| 5 |
B1 |
1589 |
1518 |
6.97 |
31.30 |
0.75 |
0.86 |
| 6 |
B2 |
1183 |
1130 |
396.23 |
2.29 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 3523.5 cm
-1
Scaled (by 0.9552) Zero Point Vibrational Energy (zpe) 3365.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/Def2TZVPP
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.095 |
-0.052 |
| Si2 |
0.000 |
-1.095 |
-0.052 |
| H3 |
0.986 |
0.000 |
0.729 |
| H4 |
-0.986 |
0.000 |
0.729 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 2.1903 | 1.6679 | 1.6679 |
Si2 | 2.1903 | | 1.6679 | 1.6679 | H3 | 1.6679 | 1.6679 | | 1.9730 | H4 | 1.6679 | 1.6679 | 1.9730 | |
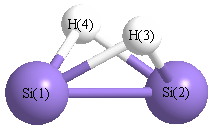 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
48.958 |
|
Si2 |
Si1 |
H3 |
48.958 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.013 |
|
|
|
| 2 |
Si |
0.013 |
|
|
|
| 3 |
H |
-0.013 |
|
|
|
| 4 |
H |
-0.013 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.363 |
0.363 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-25.709 |
0.000 |
0.000 |
| y |
0.000 |
-31.653 |
0.000 |
| z |
0.000 |
0.000 |
-28.607 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.421 |
0.000 |
0.000 |
| y |
0.000 |
-4.495 |
0.000 |
| z |
0.000 |
0.000 |
0.075 |
|
| Polar |
|---|
| 3z2-r2 | 0.149 |
|---|
| x2-y2 | 5.944 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.271 |
0.000 |
0.000 |
| y |
0.000 |
13.360 |
0.000 |
| z |
0.000 |
0.000 |
7.064 |
<r2> (average value of r
2) Å
2
| <r2> |
54.565 |
| (<r2>)1/2 |
7.387 |
Jump to
S1C1
S1C2
S1C3
Energy calculated at wB97X-D/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -580.077210 |
| Energy at 298.15K | |
| HF Energy | -580.077210 |
| Nuclear repulsion energy | 65.840734 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
2195 |
2097 |
77.53 |
289.60 |
0.36 |
0.53 |
| 2 |
A' |
1711 |
1634 |
49.62 |
83.74 |
0.18 |
0.30 |
| 3 |
A' |
1161 |
1109 |
130.77 |
6.54 |
0.44 |
0.61 |
| 4 |
A' |
650 |
621 |
21.22 |
80.57 |
0.21 |
0.35 |
| 5 |
A' |
467 |
446 |
4.23 |
2.86 |
0.19 |
0.33 |
| 6 |
A" |
159 |
152 |
36.78 |
7.30 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 3171.8 cm
-1
Scaled (by 0.9552) Zero Point Vibrational Energy (zpe) 3029.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/Def2TZVPP
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.060 |
-1.128 |
0.000 |
| Si2 |
0.060 |
0.961 |
0.000 |
| H3 |
-1.241 |
-0.022 |
0.000 |
| H4 |
-0.435 |
2.361 |
0.000 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 2.0889 | 1.7069 | 3.5236 |
Si2 | 2.0889 | | 1.6302 | 1.4845 | H3 | 1.7069 | 1.6302 | | 2.5156 | H4 | 3.5236 | 1.4845 | 2.5156 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
160.548 |
|
Si2 |
Si1 |
H3 |
49.625 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
-0.027 |
|
|
|
| 2 |
Si |
0.003 |
|
|
|
| 3 |
H |
0.002 |
|
|
|
| 4 |
H |
0.023 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.044 |
1.093 |
0.000 |
1.093 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-26.634 |
0.304 |
0.000 |
| y |
0.304 |
-25.349 |
0.000 |
| z |
0.000 |
0.000 |
-30.753 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.417 |
0.304 |
0.000 |
| y |
0.304 |
3.344 |
0.000 |
| z |
0.000 |
0.000 |
-4.762 |
|
| Polar |
|---|
| 3z2-r2 | -9.524 |
|---|
| x2-y2 | -1.285 |
|---|
| xy | 0.304 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.155 |
-0.045 |
0.000 |
| y |
-0.045 |
14.603 |
0.000 |
| z |
0.000 |
0.000 |
7.120 |
<r2> (average value of r
2) Å
2
| <r2> |
55.366 |
| (<r2>)1/2 |
7.441 |