Jump to
S1C2
Energy calculated at wB97X-D/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -1890.567398 |
| Energy at 298.15K | -1890.566871 |
| HF Energy | -1890.567398 |
| Nuclear repulsion energy | 421.828035 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
1149 |
1094 |
0.00 |
|
|
|
| 2 |
A1 |
409 |
390 |
0.00 |
|
|
|
| 3 |
A1 |
176 |
167 |
0.00 |
|
|
|
| 4 |
B1 |
14 |
14 |
0.00 |
|
|
|
| 5 |
B2 |
741 |
705 |
166.08 |
|
|
|
| 6 |
B2 |
298 |
283 |
4.62 |
|
|
|
| 7 |
E |
938 |
893 |
332.75 |
|
|
|
| 7 |
E |
938 |
893 |
332.75 |
|
|
|
| 8 |
E |
493 |
470 |
8.71 |
|
|
|
| 8 |
E |
493 |
470 |
8.71 |
|
|
|
| 9 |
E |
99 |
95 |
1.35 |
|
|
|
| 9 |
E |
99 |
95 |
1.35 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 2923.5 cm
-1
Scaled (by 0.9523) Zero Point Vibrational Energy (zpe) 2784.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/6-31+G**
Point Group is D2d
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| B1 |
0.000 |
0.000 |
0.774 |
| B2 |
0.000 |
0.000 |
-0.774 |
| Cl3 |
0.000 |
1.495 |
1.644 |
| Cl4 |
0.000 |
-1.495 |
1.644 |
| Cl5 |
1.495 |
0.000 |
-1.644 |
| Cl6 |
-1.495 |
0.000 |
-1.644 |
Atom - Atom Distances (Å)
| |
B1 |
B2 |
Cl3 |
Cl4 |
Cl5 |
Cl6 |
| B1 | | 1.5472 | 1.7297 | 1.7297 | 2.8424 | 2.8424 |
B2 | 1.5472 | | 2.8424 | 2.8424 | 1.7297 | 1.7297 | Cl3 | 1.7297 | 2.8424 | | 2.9892 | 3.9091 | 3.9091 | Cl4 | 1.7297 | 2.8424 | 2.9892 | | 3.9091 | 3.9091 | Cl5 | 2.8424 | 1.7297 | 3.9091 | 3.9091 | | 2.9892 | Cl6 | 2.8424 | 1.7297 | 3.9091 | 3.9091 | 2.9892 | |
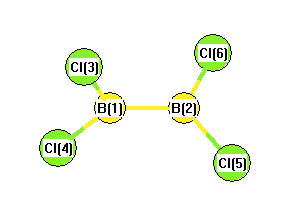 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| B1 |
B2 |
Cl5 |
120.220 |
|
B1 |
B2 |
Cl6 |
120.220 |
| B2 |
B1 |
Cl3 |
120.220 |
|
B2 |
B1 |
Cl4 |
120.220 |
| Cl3 |
B1 |
Cl4 |
119.559 |
|
Cl5 |
B2 |
Cl6 |
119.559 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
B |
-0.064 |
|
|
|
| 2 |
B |
-0.064 |
|
|
|
| 3 |
Cl |
0.032 |
|
|
|
| 4 |
Cl |
0.032 |
|
|
|
| 5 |
Cl |
0.032 |
|
|
|
| 6 |
Cl |
0.032 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-61.359 |
0.000 |
0.000 |
| y |
0.000 |
-61.359 |
0.000 |
| z |
0.000 |
0.000 |
-64.213 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.427 |
0.000 |
0.000 |
| y |
0.000 |
1.427 |
0.000 |
| z |
0.000 |
0.000 |
-2.854 |
|
| Polar |
|---|
| 3z2-r2 | -5.709 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.271 |
0.000 |
0.000 |
| y |
0.000 |
10.271 |
0.000 |
| z |
0.000 |
0.000 |
10.785 |
<r2> (average value of r
2) Å
2
| <r2> |
405.943 |
| (<r2>)1/2 |
20.148 |
Jump to
S1C1
Energy calculated at wB97X-D/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -1890.563927 |
| Energy at 298.15K | |
| HF Energy | -1890.563927 |
| Nuclear repulsion energy | 424.588341 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
1111 |
1058 |
0.00 |
|
|
|
| 2 |
Ag |
407 |
387 |
0.00 |
|
|
|
| 3 |
Ag |
193 |
184 |
0.00 |
|
|
|
| 4 |
Au |
33i |
32i |
0.00 |
|
|
|
| 5 |
B1u |
744 |
709 |
168.15 |
|
|
|
| 6 |
B1u |
303 |
289 |
3.92 |
|
|
|
| 7 |
B2g |
540 |
515 |
0.00 |
|
|
|
| 8 |
B2u |
952 |
906 |
635.25 |
|
|
|
| 9 |
B2u |
118 |
113 |
0.43 |
|
|
|
| 10 |
B3g |
976 |
930 |
0.00 |
|
|
|
| 11 |
B3g |
258 |
246 |
0.00 |
|
|
|
| 12 |
B3u |
254 |
242 |
8.64 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 2912.3 cm
-1
Scaled (by 0.9523) Zero Point Vibrational Energy (zpe) 2773.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/6-31+G**
Point Group is D2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| B1 |
0.000 |
0.000 |
0.849 |
| B2 |
0.000 |
0.000 |
-0.849 |
| Cl3 |
0.000 |
1.492 |
1.759 |
| Cl4 |
0.000 |
-1.492 |
1.759 |
| Cl5 |
0.000 |
1.492 |
-1.759 |
| Cl6 |
0.000 |
-1.492 |
-1.759 |
Atom - Atom Distances (Å)
| |
B1 |
B2 |
Cl3 |
Cl4 |
Cl5 |
Cl6 |
| B1 | | 1.6986 | 1.7475 | 1.7475 | 3.0047 | 3.0047 |
B2 | 1.6986 | | 3.0047 | 3.0047 | 1.7475 | 1.7475 | Cl3 | 1.7475 | 3.0047 | | 2.9845 | 3.5173 | 4.6129 | Cl4 | 1.7475 | 3.0047 | 2.9845 | | 4.6129 | 3.5173 | Cl5 | 3.0047 | 1.7475 | 3.5173 | 4.6129 | | 2.9845 | Cl6 | 3.0047 | 1.7475 | 4.6129 | 3.5173 | 2.9845 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| B1 |
B2 |
Cl5 |
121.357 |
|
B1 |
B2 |
Cl6 |
121.357 |
| B2 |
B1 |
Cl3 |
121.357 |
|
B2 |
B1 |
Cl4 |
121.357 |
| Cl3 |
B1 |
Cl4 |
117.287 |
|
Cl5 |
B2 |
Cl6 |
117.287 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
B |
-0.040 |
|
|
|
| 2 |
B |
-0.040 |
|
|
|
| 3 |
Cl |
0.020 |
|
|
|
| 4 |
Cl |
0.020 |
|
|
|
| 5 |
Cl |
0.020 |
|
|
|
| 6 |
Cl |
0.020 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-59.478 |
0.000 |
0.000 |
| y |
0.000 |
-62.909 |
0.000 |
| z |
0.000 |
0.000 |
-64.603 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.279 |
0.000 |
0.000 |
| y |
0.000 |
-0.869 |
0.000 |
| z |
0.000 |
0.000 |
-3.410 |
|
| Polar |
|---|
| 3z2-r2 | -6.820 |
|---|
| x2-y2 | 3.432 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.393 |
0.000 |
0.000 |
| y |
0.000 |
13.915 |
0.000 |
| z |
0.000 |
0.000 |
10.713 |
<r2> (average value of r
2) Å
2
| <r2> |
410.685 |
| (<r2>)1/2 |
20.265 |