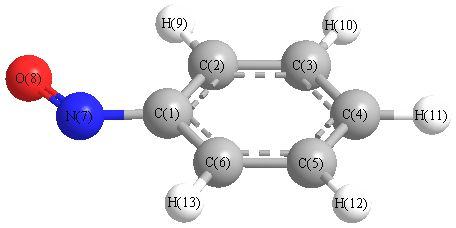
Vibrational Frequencies calculated at wB97X-D/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3252 |
3097 |
7.58 |
|
|
|
| 2 |
A' |
3247 |
3092 |
6.32 |
|
|
|
| 3 |
A' |
3238 |
3084 |
7.80 |
|
|
|
| 4 |
A' |
3232 |
3078 |
6.68 |
|
|
|
| 5 |
A' |
3219 |
3065 |
0.72 |
|
|
|
| 6 |
A' |
1704 |
1623 |
66.34 |
|
|
|
| 7 |
A' |
1680 |
1600 |
3.44 |
|
|
|
| 8 |
A' |
1643 |
1565 |
165.44 |
|
|
|
| 9 |
A' |
1522 |
1450 |
10.15 |
|
|
|
| 10 |
A' |
1507 |
1435 |
19.38 |
|
|
|
| 11 |
A' |
1375 |
1309 |
18.23 |
|
|
|
| 12 |
A' |
1344 |
1280 |
5.28 |
|
|
|
| 13 |
A' |
1214 |
1156 |
36.87 |
|
|
|
| 14 |
A' |
1195 |
1138 |
3.82 |
|
|
|
| 15 |
A' |
1159 |
1103 |
126.07 |
|
|
|
| 16 |
A' |
1107 |
1054 |
5.98 |
|
|
|
| 17 |
A' |
1048 |
998 |
7.03 |
|
|
|
| 18 |
A' |
1024 |
975 |
0.82 |
|
|
|
| 19 |
A' |
847 |
806 |
32.78 |
|
|
|
| 20 |
A' |
687 |
654 |
11.27 |
|
|
|
| 21 |
A' |
628 |
598 |
0.07 |
|
|
|
| 22 |
A' |
455 |
433 |
0.88 |
|
|
|
| 23 |
A' |
257 |
245 |
2.08 |
|
|
|
| 24 |
A" |
1027 |
978 |
0.00 |
|
|
|
| 25 |
A" |
1012 |
964 |
0.16 |
|
|
|
| 26 |
A" |
965 |
919 |
5.37 |
|
|
|
| 27 |
A" |
871 |
829 |
0.04 |
|
|
|
| 28 |
A" |
774 |
737 |
69.19 |
|
|
|
| 29 |
A" |
692 |
659 |
28.71 |
|
|
|
| 30 |
A" |
473 |
450 |
3.30 |
|
|
|
| 31 |
A" |
421 |
401 |
0.04 |
|
|
|
| 32 |
A" |
246 |
234 |
0.19 |
|
|
|
| 33 |
A" |
103 |
98 |
0.09 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 21582.8 cm
-1
Scaled (by 0.9523) Zero Point Vibrational Energy (zpe) 20553.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.567 |
|
|
|
| 2 |
C |
0.415 |
|
|
|
| 3 |
C |
-0.337 |
|
|
|
| 4 |
C |
-0.032 |
|
|
|
| 5 |
C |
-0.207 |
|
|
|
| 6 |
C |
0.058 |
|
|
|
| 7 |
N |
-0.200 |
|
|
|
| 8 |
O |
0.005 |
|
|
|
| 9 |
H |
0.185 |
|
|
|
| 10 |
H |
0.167 |
|
|
|
| 11 |
H |
0.165 |
|
|
|
| 12 |
H |
0.168 |
|
|
|
| 13 |
H |
0.180 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.164 |
-3.678 |
0.000 |
3.858 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-41.631 |
2.508 |
0.000 |
| y |
2.508 |
-48.960 |
0.000 |
| z |
0.000 |
0.000 |
-48.169 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.933 |
2.508 |
0.000 |
| y |
2.508 |
-4.060 |
0.000 |
| z |
0.000 |
0.000 |
-2.873 |
|
| Polar |
|---|
| 3z2-r2 | -5.746 |
|---|
| x2-y2 | 7.329 |
|---|
| xy | 2.508 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
13.247 |
-1.678 |
0.000 |
| y |
-1.678 |
15.783 |
0.000 |
| z |
0.000 |
0.000 |
6.413 |
<r2> (average value of r
2) Å
2
| <r2> |
249.094 |
| (<r2>)1/2 |
15.783 |