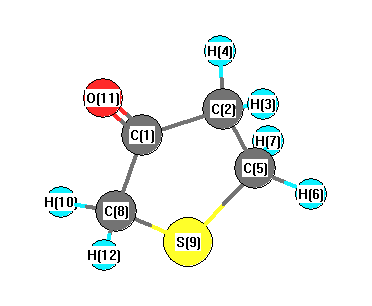
Vibrational Frequencies calculated at wB97X-D/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3176 |
3025 |
4.98 |
|
|
|
| 2 |
A |
3168 |
3018 |
0.19 |
|
|
|
| 3 |
A |
3157 |
3007 |
6.57 |
|
|
|
| 4 |
A |
3100 |
2953 |
24.67 |
|
|
|
| 5 |
A |
3091 |
2945 |
12.25 |
|
|
|
| 6 |
A |
3081 |
2935 |
5.00 |
|
|
|
| 7 |
A |
1861 |
1773 |
261.69 |
|
|
|
| 8 |
A |
1504 |
1433 |
3.58 |
|
|
|
| 9 |
A |
1459 |
1390 |
6.26 |
|
|
|
| 10 |
A |
1457 |
1388 |
17.68 |
|
|
|
| 11 |
A |
1320 |
1258 |
20.55 |
|
|
|
| 12 |
A |
1314 |
1252 |
3.69 |
|
|
|
| 13 |
A |
1253 |
1193 |
27.41 |
|
|
|
| 14 |
A |
1235 |
1177 |
2.45 |
|
|
|
| 15 |
A |
1182 |
1126 |
7.26 |
|
|
|
| 16 |
A |
1165 |
1110 |
46.83 |
|
|
|
| 17 |
A |
1112 |
1059 |
2.50 |
|
|
|
| 18 |
A |
1012 |
964 |
7.51 |
|
|
|
| 19 |
A |
994 |
947 |
0.85 |
|
|
|
| 20 |
A |
884 |
842 |
6.56 |
|
|
|
| 21 |
A |
842 |
802 |
1.94 |
|
|
|
| 22 |
A |
806 |
767 |
0.68 |
|
|
|
| 23 |
A |
740 |
705 |
6.66 |
|
|
|
| 24 |
A |
696 |
663 |
0.95 |
|
|
|
| 25 |
A |
568 |
541 |
4.29 |
|
|
|
| 26 |
A |
494 |
471 |
5.48 |
|
|
|
| 27 |
A |
451 |
430 |
4.92 |
|
|
|
| 28 |
A |
436 |
416 |
4.54 |
|
|
|
| 29 |
A |
181 |
173 |
2.29 |
|
|
|
| 30 |
A |
75 |
72 |
13.36 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20907.1 cm
-1
Scaled (by 0.9526) Zero Point Vibrational Energy (zpe) 19916.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.130 |
|
|
|
| 2 |
C |
-0.179 |
|
|
|
| 3 |
H |
0.197 |
|
|
|
| 4 |
H |
0.208 |
|
|
|
| 5 |
C |
-0.467 |
|
|
|
| 6 |
H |
0.200 |
|
|
|
| 7 |
H |
0.199 |
|
|
|
| 8 |
C |
-0.350 |
|
|
|
| 9 |
S |
0.022 |
|
|
|
| 10 |
H |
0.222 |
|
|
|
| 11 |
O |
-0.403 |
|
|
|
| 12 |
H |
0.221 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.381 |
1.412 |
0.420 |
2.019 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-55.055 |
1.048 |
0.407 |
| y |
1.048 |
-38.290 |
-0.038 |
| z |
0.407 |
-0.038 |
-42.268 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-14.776 |
1.048 |
0.407 |
| y |
1.048 |
10.372 |
-0.038 |
| z |
0.407 |
-0.038 |
4.404 |
|
| Polar |
|---|
| 3z2-r2 | 8.808 |
|---|
| x2-y2 | -16.765 |
|---|
| xy | 1.048 |
|---|
| xz | 0.407 |
|---|
| yz | -0.038 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.853 |
0.082 |
-0.082 |
| y |
0.082 |
9.828 |
-0.050 |
| z |
-0.082 |
-0.050 |
7.556 |
<r2> (average value of r
2) Å
2
| <r2> |
185.793 |
| (<r2>)1/2 |
13.631 |