Vibrational Frequencies calculated at wB97X-D/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3864 |
3681 |
93.14 |
|
|
|
| 2 |
A' |
3259 |
3104 |
1.35 |
|
|
|
| 3 |
A' |
3253 |
3098 |
12.43 |
|
|
|
| 4 |
A' |
3226 |
3073 |
10.35 |
|
|
|
| 5 |
A' |
3217 |
3064 |
11.51 |
|
|
|
| 6 |
A' |
1692 |
1612 |
113.58 |
|
|
|
| 7 |
A' |
1670 |
1591 |
104.44 |
|
|
|
| 8 |
A' |
1541 |
1468 |
161.23 |
|
|
|
| 9 |
A' |
1510 |
1438 |
38.97 |
|
|
|
| 10 |
A' |
1384 |
1318 |
47.11 |
|
|
|
| 11 |
A' |
1357 |
1293 |
49.00 |
|
|
|
| 12 |
A' |
1335 |
1271 |
9.26 |
|
|
|
| 13 |
A' |
1217 |
1159 |
149.90 |
|
|
|
| 14 |
A' |
1175 |
1120 |
29.15 |
|
|
|
| 15 |
A' |
1122 |
1069 |
24.62 |
|
|
|
| 16 |
A' |
1078 |
1027 |
3.92 |
|
|
|
| 17 |
A' |
1018 |
970 |
6.88 |
|
|
|
| 18 |
A' |
876 |
835 |
11.79 |
|
|
|
| 19 |
A' |
643 |
613 |
2.30 |
|
|
|
| 20 |
A' |
573 |
546 |
0.43 |
|
|
|
| 21 |
A' |
427 |
406 |
18.02 |
|
|
|
| 22 |
A" |
1018 |
970 |
0.03 |
|
|
|
| 23 |
A" |
984 |
937 |
0.54 |
|
|
|
| 24 |
A" |
888 |
846 |
3.73 |
|
|
|
| 25 |
A" |
797 |
760 |
63.84 |
|
|
|
| 26 |
A" |
756 |
720 |
9.98 |
|
|
|
| 27 |
A" |
561 |
535 |
28.67 |
|
|
|
| 28 |
A" |
500 |
477 |
108.19 |
|
|
|
| 29 |
A" |
424 |
404 |
0.58 |
|
|
|
| 30 |
A" |
217 |
207 |
1.19 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20790.9 cm
-1
Scaled (by 0.9526) Zero Point Vibrational Energy (zpe) 19805.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
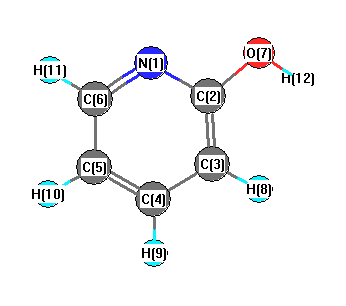
Charges from optimized geometry at wB97X-D/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.313 |
|
|
|
| 2 |
C |
0.122 |
|
|
|
| 3 |
C |
0.194 |
|
|
|
| 4 |
C |
-0.308 |
|
|
|
| 5 |
C |
-0.019 |
|
|
|
| 6 |
C |
-0.222 |
|
|
|
| 7 |
O |
-0.489 |
|
|
|
| 8 |
H |
0.174 |
|
|
|
| 9 |
H |
0.166 |
|
|
|
| 10 |
H |
0.162 |
|
|
|
| 11 |
H |
0.153 |
|
|
|
| 12 |
H |
0.377 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.512 |
-1.388 |
0.000 |
1.479 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-37.567 |
-2.164 |
0.000 |
| y |
-2.164 |
-36.516 |
0.000 |
| z |
0.000 |
0.000 |
-43.380 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
2.381 |
-2.164 |
0.000 |
| y |
-2.164 |
3.957 |
0.000 |
| z |
0.000 |
0.000 |
-6.338 |
|
| Polar |
|---|
| 3z2-r2 | -12.677 |
|---|
| x2-y2 | -1.051 |
|---|
| xy | -2.164 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.857 |
0.015 |
0.000 |
| y |
0.015 |
12.407 |
0.000 |
| z |
0.000 |
0.000 |
5.705 |
<r2> (average value of r
2) Å
2
| <r2> |
176.282 |
| (<r2>)1/2 |
13.277 |