Jump to
S1C2
Energy calculated at wB97X-D/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -339.538130 |
| Energy at 298.15K | -339.547119 |
| HF Energy | -339.538130 |
| Nuclear repulsion energy | 259.964410 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3237 |
3084 |
0.00 |
|
|
|
| 2 |
Ag |
3091 |
2944 |
0.00 |
|
|
|
| 3 |
Ag |
1623 |
1546 |
0.00 |
|
|
|
| 4 |
Ag |
1488 |
1417 |
0.00 |
|
|
|
| 5 |
Ag |
1464 |
1395 |
0.00 |
|
|
|
| 6 |
Ag |
1430 |
1362 |
0.00 |
|
|
|
| 7 |
Ag |
1118 |
1065 |
0.00 |
|
|
|
| 8 |
Ag |
819 |
780 |
0.00 |
|
|
|
| 9 |
Ag |
639 |
609 |
0.00 |
|
|
|
| 10 |
Ag |
416 |
396 |
0.00 |
|
|
|
| 11 |
Au |
3177 |
3026 |
4.70 |
|
|
|
| 12 |
Au |
1474 |
1404 |
30.64 |
|
|
|
| 13 |
Au |
1140 |
1086 |
0.07 |
|
|
|
| 14 |
Au |
338 |
322 |
6.36 |
|
|
|
| 15 |
Au |
184 |
175 |
16.78 |
|
|
|
| 16 |
Au |
77 |
74 |
2.21 |
|
|
|
| 17 |
Bg |
3176 |
3026 |
0.00 |
|
|
|
| 18 |
Bg |
1471 |
1401 |
0.00 |
|
|
|
| 19 |
Bg |
1116 |
1063 |
0.00 |
|
|
|
| 20 |
Bg |
527 |
502 |
0.00 |
|
|
|
| 21 |
Bg |
123 |
117 |
0.00 |
|
|
|
| 22 |
Bu |
3237 |
3084 |
0.96 |
|
|
|
| 23 |
Bu |
3091 |
2944 |
11.27 |
|
|
|
| 24 |
Bu |
1485 |
1414 |
4.84 |
|
|
|
| 25 |
Bu |
1445 |
1377 |
42.70 |
|
|
|
| 26 |
Bu |
1371 |
1306 |
465.42 |
|
|
|
| 27 |
Bu |
1173 |
1117 |
55.11 |
|
|
|
| 28 |
Bu |
982 |
935 |
53.64 |
|
|
|
| 29 |
Bu |
557 |
531 |
40.93 |
|
|
|
| 30 |
Bu |
304 |
289 |
25.62 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20884.6 cm
-1
Scaled (by 0.9526) Zero Point Vibrational Energy (zpe) 19894.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/cc-pVDZ
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
-0.060 |
0.646 |
0.000 |
| N2 |
0.060 |
-0.646 |
0.000 |
| O3 |
-1.187 |
1.229 |
0.000 |
| O4 |
1.187 |
-1.229 |
0.000 |
| C5 |
1.187 |
1.393 |
0.000 |
| C6 |
-1.187 |
-1.393 |
0.000 |
| H7 |
0.893 |
2.440 |
0.000 |
| H8 |
1.767 |
1.137 |
0.890 |
| H9 |
1.767 |
1.137 |
-0.890 |
| H10 |
-0.893 |
-2.440 |
0.000 |
| H11 |
-1.767 |
-1.137 |
0.890 |
| H12 |
-1.767 |
-1.137 |
-0.890 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
| N1 | | 1.2969 | 1.2698 | 2.2517 | 1.4540 | 2.3301 | 2.0314 | 2.0907 | 2.0907 | 3.1962 | 2.6242 | 2.6242 |
N2 | 1.2969 | | 2.2517 | 1.2698 | 2.3301 | 1.4540 | 3.1962 | 2.6242 | 2.6242 | 2.0314 | 2.0907 | 2.0907 | O3 | 1.2698 | 2.2517 | | 3.4181 | 2.3806 | 2.6224 | 2.4071 | 3.0872 | 3.0872 | 3.6809 | 2.5938 | 2.5938 | O4 | 2.2517 | 1.2698 | 3.4181 | | 2.6224 | 2.3806 | 3.6809 | 2.5938 | 2.5938 | 2.4071 | 3.0872 | 3.0872 | C5 | 1.4540 | 2.3301 | 2.3806 | 2.6224 | | 3.6613 | 1.0874 | 1.0926 | 1.0926 | 4.3613 | 3.9906 | 3.9906 | C6 | 2.3301 | 1.4540 | 2.6224 | 2.3806 | 3.6613 | | 4.3613 | 3.9906 | 3.9906 | 1.0874 | 1.0926 | 1.0926 | H7 | 2.0314 | 3.1962 | 2.4071 | 3.6809 | 1.0874 | 4.3613 | | 1.8038 | 1.8038 | 5.1964 | 4.5458 | 4.5458 | H8 | 2.0907 | 2.6242 | 3.0872 | 2.5938 | 1.0926 | 3.9906 | 1.8038 | | 1.7799 | 4.5458 | 4.2030 | 4.5643 | H9 | 2.0907 | 2.6242 | 3.0872 | 2.5938 | 1.0926 | 3.9906 | 1.8038 | 1.7799 | | 4.5458 | 4.5643 | 4.2030 | H10 | 3.1962 | 2.0314 | 3.6809 | 2.4071 | 4.3613 | 1.0874 | 5.1964 | 4.5458 | 4.5458 | | 1.8038 | 1.8038 | H11 | 2.6242 | 2.0907 | 2.5938 | 3.0872 | 3.9906 | 1.0926 | 4.5458 | 4.2030 | 4.5643 | 1.8038 | | 1.7799 | H12 | 2.6242 | 2.0907 | 2.5938 | 3.0872 | 3.9906 | 1.0926 | 4.5458 | 4.5643 | 4.2030 | 1.8038 | 1.7799 | |
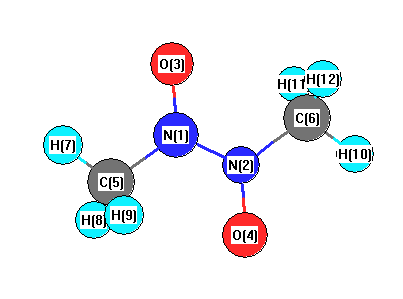 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
N2 |
O4 |
122.629 |
|
N1 |
N2 |
C6 |
115.663 |
| N1 |
C5 |
H7 |
105.222 |
|
N1 |
C5 |
H8 |
109.550 |
| N1 |
C5 |
H9 |
109.550 |
|
N2 |
N1 |
O3 |
122.629 |
| N2 |
N1 |
C5 |
115.663 |
|
N2 |
C6 |
H10 |
105.222 |
| N2 |
C6 |
H11 |
109.550 |
|
N2 |
C6 |
H12 |
109.550 |
| O3 |
N1 |
C5 |
121.708 |
|
O4 |
N2 |
C6 |
121.708 |
| H7 |
C5 |
H8 |
111.675 |
|
H7 |
C5 |
H9 |
111.675 |
| H8 |
C5 |
H9 |
109.089 |
|
H10 |
C6 |
H11 |
111.675 |
| H10 |
C6 |
H12 |
111.675 |
|
H11 |
C6 |
H12 |
109.089 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
0.042 |
|
|
|
| 2 |
N |
0.042 |
|
|
|
| 3 |
O |
-0.277 |
|
|
|
| 4 |
O |
-0.277 |
|
|
|
| 5 |
C |
-0.369 |
|
|
|
| 6 |
C |
-0.369 |
|
|
|
| 7 |
H |
0.199 |
|
|
|
| 8 |
H |
0.202 |
|
|
|
| 9 |
H |
0.202 |
|
|
|
| 10 |
H |
0.199 |
|
|
|
| 11 |
H |
0.202 |
|
|
|
| 12 |
H |
0.202 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-37.886 |
11.184 |
0.000 |
| y |
11.184 |
-35.770 |
0.000 |
| z |
0.000 |
0.000 |
-34.746 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.628 |
11.184 |
0.000 |
| y |
11.184 |
0.546 |
0.000 |
| z |
0.000 |
0.000 |
2.082 |
|
| Polar |
|---|
| 3z2-r2 | 4.165 |
|---|
| x2-y2 | -2.116 |
|---|
| xy | 11.184 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.509 |
-1.083 |
0.000 |
| y |
-1.083 |
10.090 |
0.000 |
| z |
0.000 |
0.000 |
5.302 |
<r2> (average value of r
2) Å
2
| <r2> |
149.817 |
| (<r2>)1/2 |
12.240 |
Jump to
S1C1
Energy calculated at wB97X-D/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -339.538196 |
| Energy at 298.15K | -339.547546 |
| HF Energy | -339.538196 |
| Nuclear repulsion energy | 259.981936 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3242 |
3089 |
0.00 |
|
|
|
| 2 |
Ag |
3180 |
3029 |
0.00 |
|
|
|
| 3 |
Ag |
3094 |
2948 |
0.00 |
|
|
|
| 4 |
Ag |
1625 |
1548 |
0.00 |
|
|
|
| 5 |
Ag |
1490 |
1419 |
0.00 |
|
|
|
| 6 |
Ag |
1481 |
1411 |
0.00 |
|
|
|
| 7 |
Ag |
1478 |
1408 |
0.00 |
|
|
|
| 8 |
Ag |
1436 |
1368 |
0.00 |
|
|
|
| 9 |
Ag |
1125 |
1072 |
0.00 |
|
|
|
| 10 |
Ag |
1124 |
1071 |
0.00 |
|
|
|
| 11 |
Ag |
819 |
780 |
0.00 |
|
|
|
| 12 |
Ag |
639 |
609 |
0.00 |
|
|
|
| 13 |
Ag |
528 |
502 |
0.00 |
|
|
|
| 14 |
Ag |
419 |
399 |
0.00 |
|
|
|
| 15 |
Ag |
218 |
208 |
0.00 |
|
|
|
| 16 |
Au |
3243 |
3089 |
0.97 |
|
|
|
| 17 |
Au |
3180 |
3029 |
4.69 |
|
|
|
| 18 |
Au |
3094 |
2948 |
11.20 |
|
|
|
| 19 |
Au |
1499 |
1428 |
0.92 |
|
|
|
| 20 |
Au |
1484 |
1413 |
30.30 |
|
|
|
| 21 |
Au |
1451 |
1382 |
45.20 |
|
|
|
| 22 |
Au |
1373 |
1308 |
460.32 |
|
|
|
| 23 |
Au |
1176 |
1120 |
61.24 |
|
|
|
| 24 |
Au |
1149 |
1094 |
0.04 |
|
|
|
| 25 |
Au |
985 |
938 |
53.09 |
|
|
|
| 26 |
Au |
558 |
531 |
40.96 |
|
|
|
| 27 |
Au |
339 |
323 |
6.35 |
|
|
|
| 28 |
Au |
308 |
293 |
25.62 |
|
|
|
| 29 |
Au |
218 |
208 |
3.21 |
|
|
|
| 30 |
Au |
162 |
154 |
15.82 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 21057.4 cm
-1
Scaled (by 0.9526) Zero Point Vibrational Energy (zpe) 20059.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/cc-pVDZ
Point Group is Ci
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
-0.633 |
0.358 |
0.134 |
| N2 |
0.633 |
-0.358 |
-0.134 |
| O3 |
-0.678 |
1.615 |
-0.083 |
| O4 |
0.678 |
-1.615 |
0.083 |
| C5 |
-1.822 |
-0.544 |
-0.007 |
| C6 |
1.822 |
0.544 |
0.007 |
| H7 |
-2.376 |
-0.275 |
-0.915 |
| H8 |
-1.381 |
-1.554 |
-0.057 |
| H9 |
-2.448 |
-0.425 |
0.884 |
| H10 |
2.448 |
0.425 |
-0.884 |
| H11 |
1.381 |
1.554 |
0.057 |
| H12 |
2.376 |
0.275 |
0.915 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
| N1 | | 1.4782 | 1.2768 | 2.3692 | 1.4987 | 2.4647 | 2.1300 | 2.0612 | 2.1146 | 3.2456 | 2.3432 | 3.1092 |
N2 | 1.4782 | | 2.3692 | 1.2768 | 2.4647 | 1.4987 | 3.1092 | 2.3432 | 3.2456 | 2.1146 | 2.0612 | 2.1300 | O3 | 1.2768 | 2.3692 | | 3.5074 | 2.4443 | 2.7212 | 2.6729 | 3.2457 | 2.8688 | 3.4400 | 2.0647 | 3.4811 | O4 | 2.3692 | 1.2768 | 3.5074 | | 2.7212 | 2.4443 | 3.4811 | 2.0647 | 3.4400 | 2.8688 | 3.2457 | 2.6729 | C5 | 1.4987 | 2.4647 | 2.4443 | 2.7212 | | 3.8022 | 1.0969 | 1.1028 | 1.0960 | 4.4654 | 3.8287 | 4.3746 | C6 | 2.4647 | 1.4987 | 2.7212 | 2.4443 | 3.8022 | | 4.3746 | 3.8287 | 4.4654 | 1.0960 | 1.1028 | 1.0969 | H7 | 2.1300 | 3.1092 | 2.6729 | 3.4811 | 1.0969 | 4.3746 | | 1.8333 | 1.8066 | 4.8745 | 4.2892 | 5.1209 | H8 | 2.0612 | 2.3432 | 3.2457 | 2.0647 | 1.1028 | 3.8287 | 1.8333 | | 1.8163 | 4.3887 | 4.1585 | 4.2892 | H9 | 2.1146 | 3.2456 | 2.8688 | 3.4400 | 1.0960 | 4.4654 | 1.8066 | 1.8163 | | 5.2750 | 4.3887 | 4.8745 | H10 | 3.2456 | 2.1146 | 3.4400 | 2.8688 | 4.4654 | 1.0960 | 4.8745 | 4.3887 | 5.2750 | | 1.8163 | 1.8066 | H11 | 2.3432 | 2.0612 | 2.0647 | 3.2457 | 3.8287 | 1.1028 | 4.2892 | 4.1585 | 4.3887 | 1.8163 | | 1.8333 | H12 | 3.1092 | 2.1300 | 3.4811 | 2.6729 | 4.3746 | 1.0969 | 5.1209 | 4.2892 | 4.8745 | 1.8066 | 1.8333 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
N2 |
O4 |
118.445 |
|
N1 |
N2 |
C6 |
111.776 |
| N1 |
C5 |
H7 |
109.315 |
|
N1 |
C5 |
H8 |
103.764 |
| N1 |
C5 |
H9 |
108.164 |
|
N2 |
N1 |
O3 |
118.445 |
| N2 |
N1 |
C5 |
111.776 |
|
N2 |
C6 |
H10 |
108.164 |
| N2 |
C6 |
H11 |
103.764 |
|
N2 |
C6 |
H12 |
109.315 |
| O3 |
N1 |
C5 |
123.251 |
|
O4 |
N2 |
C6 |
123.251 |
| H7 |
C5 |
H8 |
112.908 |
|
H7 |
C5 |
H9 |
110.942 |
| H8 |
C5 |
H9 |
111.392 |
|
H10 |
C6 |
H11 |
111.392 |
| H10 |
C6 |
H12 |
110.942 |
|
H11 |
C6 |
H12 |
112.908 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
0.042 |
|
|
|
| 2 |
N |
0.042 |
|
|
|
| 3 |
O |
-0.277 |
|
|
|
| 4 |
O |
-0.277 |
|
|
|
| 5 |
C |
-0.368 |
|
|
|
| 6 |
C |
-0.368 |
|
|
|
| 7 |
H |
0.199 |
|
|
|
| 8 |
H |
0.202 |
|
|
|
| 9 |
H |
0.202 |
|
|
|
| 10 |
H |
0.202 |
|
|
|
| 11 |
H |
0.202 |
|
|
|
| 12 |
H |
0.199 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-27.728 |
6.027 |
-1.540 |
| y |
6.027 |
-42.790 |
5.458 |
| z |
-1.540 |
5.458 |
-37.888 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
12.610 |
6.027 |
-1.540 |
| y |
6.027 |
-9.981 |
5.458 |
| z |
-1.540 |
5.458 |
-2.629 |
|
| Polar |
|---|
| 3z2-r2 | -5.257 |
|---|
| x2-y2 | 15.061 |
|---|
| xy | 6.027 |
|---|
| xz | -1.540 |
|---|
| yz | 5.458 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.796 |
-0.816 |
1.192 |
| y |
-0.816 |
8.562 |
-1.856 |
| z |
1.192 |
-1.856 |
6.513 |
<r2> (average value of r
2) Å
2
| <r2> |
149.802 |
| (<r2>)1/2 |
12.239 |