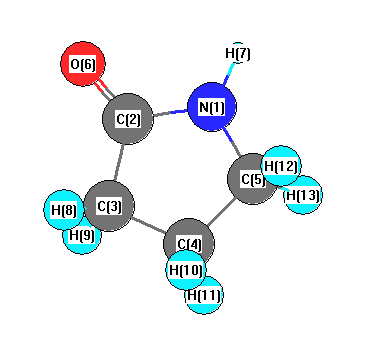
Vibrational Frequencies calculated at wB97X-D/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3699 |
3524 |
41.03 |
|
|
|
| 2 |
A |
3173 |
3023 |
12.26 |
|
|
|
| 3 |
A |
3156 |
3007 |
20.21 |
|
|
|
| 4 |
A |
3120 |
2972 |
35.38 |
|
|
|
| 5 |
A |
3098 |
2951 |
19.43 |
|
|
|
| 6 |
A |
3093 |
2946 |
13.45 |
|
|
|
| 7 |
A |
3039 |
2895 |
64.41 |
|
|
|
| 8 |
A |
1842 |
1754 |
560.70 |
|
|
|
| 9 |
A |
1552 |
1479 |
2.96 |
|
|
|
| 10 |
A |
1516 |
1445 |
12.17 |
|
|
|
| 11 |
A |
1479 |
1409 |
24.53 |
|
|
|
| 12 |
A |
1472 |
1403 |
32.77 |
|
|
|
| 13 |
A |
1391 |
1325 |
8.90 |
|
|
|
| 14 |
A |
1363 |
1298 |
19.85 |
|
|
|
| 15 |
A |
1327 |
1264 |
51.74 |
|
|
|
| 16 |
A |
1290 |
1229 |
60.19 |
|
|
|
| 17 |
A |
1264 |
1204 |
3.86 |
|
|
|
| 18 |
A |
1229 |
1171 |
1.81 |
|
|
|
| 19 |
A |
1201 |
1144 |
5.20 |
|
|
|
| 20 |
A |
1109 |
1056 |
9.40 |
|
|
|
| 21 |
A |
1098 |
1046 |
9.10 |
|
|
|
| 22 |
A |
1021 |
972 |
11.26 |
|
|
|
| 23 |
A |
937 |
892 |
0.57 |
|
|
|
| 24 |
A |
927 |
883 |
1.23 |
|
|
|
| 25 |
A |
903 |
860 |
4.49 |
|
|
|
| 26 |
A |
829 |
789 |
4.70 |
|
|
|
| 27 |
A |
702 |
669 |
6.50 |
|
|
|
| 28 |
A |
643 |
613 |
19.83 |
|
|
|
| 29 |
A |
568 |
541 |
46.68 |
|
|
|
| 30 |
A |
486 |
463 |
66.13 |
|
|
|
| 31 |
A |
472 |
449 |
15.94 |
|
|
|
| 32 |
A |
199 |
190 |
5.10 |
|
|
|
| 33 |
A |
140 |
133 |
0.79 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 24669.0 cm
-1
Scaled (by 0.9526) Zero Point Vibrational Energy (zpe) 23499.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.388 |
|
|
|
| 2 |
C |
0.271 |
|
|
|
| 3 |
C |
-0.164 |
|
|
|
| 4 |
C |
-0.315 |
|
|
|
| 5 |
C |
-0.220 |
|
|
|
| 6 |
O |
-0.543 |
|
|
|
| 7 |
H |
0.324 |
|
|
|
| 8 |
H |
0.190 |
|
|
|
| 9 |
H |
0.190 |
|
|
|
| 10 |
H |
0.165 |
|
|
|
| 11 |
H |
0.176 |
|
|
|
| 12 |
H |
0.156 |
|
|
|
| 13 |
H |
0.158 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-4.398 |
-0.576 |
0.402 |
4.454 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-44.721 |
-0.214 |
0.193 |
| y |
-0.214 |
-31.333 |
-0.248 |
| z |
0.193 |
-0.248 |
-35.721 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-11.193 |
-0.214 |
0.193 |
| y |
-0.214 |
8.888 |
-0.248 |
| z |
0.193 |
-0.248 |
2.305 |
|
| Polar |
|---|
| 3z2-r2 | 4.611 |
|---|
| x2-y2 | -13.388 |
|---|
| xy | -0.214 |
|---|
| xz | 0.193 |
|---|
| yz | -0.248 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.475 |
0.441 |
-0.075 |
| y |
0.441 |
8.088 |
0.061 |
| z |
-0.075 |
0.061 |
6.336 |
<r2> (average value of r
2) Å
2
| <r2> |
146.100 |
| (<r2>)1/2 |
12.087 |