Vibrational Frequencies calculated at wB97X-D/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3157 |
3008 |
25.49 |
|
|
|
| 2 |
A' |
3136 |
2987 |
13.79 |
|
|
|
| 3 |
A' |
3073 |
2928 |
20.31 |
|
|
|
| 4 |
A' |
3010 |
2867 |
60.19 |
|
|
|
| 5 |
A' |
2996 |
2854 |
38.84 |
|
|
|
| 6 |
A' |
1532 |
1459 |
3.22 |
|
|
|
| 7 |
A' |
1514 |
1442 |
3.63 |
|
|
|
| 8 |
A' |
1502 |
1431 |
5.40 |
|
|
|
| 9 |
A' |
1496 |
1425 |
4.73 |
|
|
|
| 10 |
A' |
1461 |
1391 |
10.48 |
|
|
|
| 11 |
A' |
1418 |
1351 |
26.16 |
|
|
|
| 12 |
A' |
1391 |
1325 |
12.68 |
|
|
|
| 13 |
A' |
1298 |
1237 |
20.04 |
|
|
|
| 14 |
A' |
1204 |
1147 |
248.13 |
|
|
|
| 15 |
A' |
1166 |
1110 |
21.50 |
|
|
|
| 16 |
A' |
1090 |
1038 |
4.38 |
|
|
|
| 17 |
A' |
1067 |
1016 |
15.79 |
|
|
|
| 18 |
A' |
915 |
871 |
12.82 |
|
|
|
| 19 |
A' |
782 |
745 |
46.75 |
|
|
|
| 20 |
A' |
487 |
464 |
0.64 |
|
|
|
| 21 |
A' |
385 |
367 |
2.54 |
|
|
|
| 22 |
A' |
270 |
257 |
3.33 |
|
|
|
| 23 |
A' |
122 |
116 |
1.36 |
|
|
|
| 24 |
A" |
3207 |
3055 |
4.70 |
|
|
|
| 25 |
A" |
3166 |
3016 |
25.75 |
|
|
|
| 26 |
A" |
3054 |
2909 |
27.74 |
|
|
|
| 27 |
A" |
3034 |
2890 |
66.97 |
|
|
|
| 28 |
A" |
1490 |
1420 |
8.32 |
|
|
|
| 29 |
A" |
1313 |
1250 |
4.23 |
|
|
|
| 30 |
A" |
1302 |
1241 |
0.38 |
|
|
|
| 31 |
A" |
1222 |
1164 |
4.73 |
|
|
|
| 32 |
A" |
1179 |
1123 |
4.45 |
|
|
|
| 33 |
A" |
1066 |
1015 |
2.40 |
|
|
|
| 34 |
A" |
818 |
779 |
0.65 |
|
|
|
| 35 |
A" |
799 |
761 |
0.12 |
|
|
|
| 36 |
A" |
226 |
215 |
1.30 |
|
|
|
| 37 |
A" |
141 |
135 |
9.09 |
|
|
|
| 38 |
A" |
56 |
54 |
0.01 |
|
|
|
| 39 |
A" |
54i |
51i |
2.37 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 28244.0 cm
-1
Scaled (by 0.9526) Zero Point Vibrational Energy (zpe) 26905.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
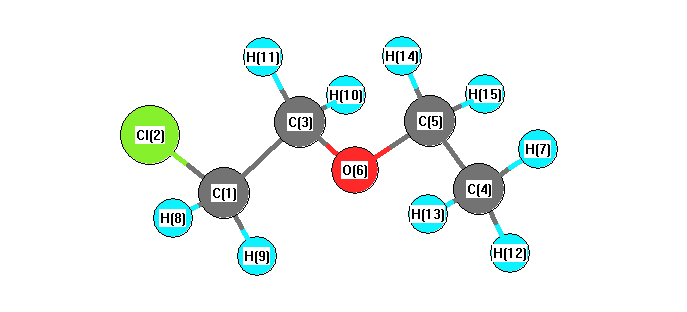
Charges from optimized geometry at wB97X-D/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.402 |
|
|
|
| 2 |
Cl |
-0.041 |
|
|
|
| 3 |
C |
-0.175 |
|
|
|
| 4 |
C |
-0.604 |
|
|
|
| 5 |
C |
0.089 |
|
|
|
| 6 |
O |
-0.355 |
|
|
|
| 7 |
H |
0.159 |
|
|
|
| 8 |
H |
0.214 |
|
|
|
| 9 |
H |
0.214 |
|
|
|
| 10 |
H |
0.148 |
|
|
|
| 11 |
H |
0.148 |
|
|
|
| 12 |
H |
0.173 |
|
|
|
| 13 |
H |
0.173 |
|
|
|
| 14 |
H |
0.130 |
|
|
|
| 15 |
H |
0.130 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.227 |
-0.194 |
0.000 |
2.236 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.426 |
3.446 |
0.000 |
| y |
3.446 |
-46.495 |
0.000 |
| z |
0.000 |
0.000 |
-44.417 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.970 |
3.446 |
0.000 |
| y |
3.446 |
-1.074 |
0.000 |
| z |
0.000 |
0.000 |
2.044 |
|
| Polar |
|---|
| 3z2-r2 | 4.087 |
|---|
| x2-y2 | 0.070 |
|---|
| xy | 3.446 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.475 |
-1.351 |
0.000 |
| y |
-1.351 |
9.056 |
0.000 |
| z |
0.000 |
0.000 |
7.907 |
<r2> (average value of r
2) Å
2
| <r2> |
365.778 |
| (<r2>)1/2 |
19.125 |