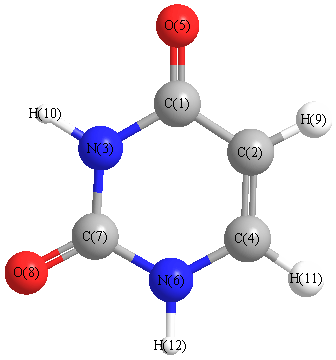
Vibrational Frequencies calculated at wB97X-D/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3699 |
3524 |
116.23 |
|
|
|
| 2 |
A' |
3660 |
3486 |
75.97 |
|
|
|
| 3 |
A' |
3297 |
3141 |
1.47 |
|
|
|
| 4 |
A' |
3256 |
3101 |
2.71 |
|
|
|
| 5 |
A' |
1858 |
1770 |
644.18 |
|
|
|
| 6 |
A' |
1827 |
1740 |
900.99 |
|
|
|
| 7 |
A' |
1717 |
1636 |
73.27 |
|
|
|
| 8 |
A' |
1532 |
1460 |
135.39 |
|
|
|
| 9 |
A' |
1443 |
1374 |
112.48 |
|
|
|
| 10 |
A' |
1433 |
1365 |
17.49 |
|
|
|
| 11 |
A' |
1407 |
1340 |
8.06 |
|
|
|
| 12 |
A' |
1258 |
1198 |
2.70 |
|
|
|
| 13 |
A' |
1228 |
1170 |
110.87 |
|
|
|
| 14 |
A' |
1111 |
1058 |
8.12 |
|
|
|
| 15 |
A' |
1000 |
953 |
7.15 |
|
|
|
| 16 |
A' |
987 |
940 |
8.16 |
|
|
|
| 17 |
A' |
784 |
747 |
3.23 |
|
|
|
| 18 |
A' |
566 |
539 |
4.85 |
|
|
|
| 19 |
A' |
550 |
524 |
5.29 |
|
|
|
| 20 |
A' |
525 |
500 |
20.19 |
|
|
|
| 21 |
A' |
392 |
374 |
23.46 |
|
|
|
| 22 |
A" |
978 |
932 |
0.43 |
|
|
|
| 23 |
A" |
816 |
777 |
59.35 |
|
|
|
| 24 |
A" |
756 |
720 |
56.76 |
|
|
|
| 25 |
A" |
730 |
696 |
8.99 |
|
|
|
| 26 |
A" |
679 |
646 |
92.10 |
|
|
|
| 27 |
A" |
567 |
540 |
48.41 |
|
|
|
| 28 |
A" |
393 |
375 |
27.55 |
|
|
|
| 29 |
A" |
165 |
157 |
0.28 |
|
|
|
| 30 |
A" |
148 |
141 |
0.91 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 19380.1 cm
-1
Scaled (by 0.9526) Zero Point Vibrational Energy (zpe) 18461.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.289 |
|
|
|
| 2 |
C |
0.107 |
|
|
|
| 3 |
N |
-0.579 |
|
|
|
| 4 |
C |
-0.103 |
|
|
|
| 5 |
O |
-0.508 |
|
|
|
| 6 |
N |
-0.471 |
|
|
|
| 7 |
C |
0.693 |
|
|
|
| 8 |
O |
-0.525 |
|
|
|
| 9 |
H |
0.192 |
|
|
|
| 10 |
H |
0.363 |
|
|
|
| 11 |
H |
0.187 |
|
|
|
| 12 |
H |
0.356 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.165 |
-4.530 |
0.000 |
4.678 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-58.630 |
1.671 |
0.000 |
| y |
1.671 |
-37.846 |
0.000 |
| z |
0.000 |
0.000 |
-46.518 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-16.448 |
1.671 |
0.000 |
| y |
1.671 |
14.729 |
0.000 |
| z |
0.000 |
0.000 |
1.720 |
|
| Polar |
|---|
| 3z2-r2 | 3.439 |
|---|
| x2-y2 | -20.785 |
|---|
| xy | 1.671 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
13.215 |
0.500 |
0.000 |
| y |
0.500 |
10.226 |
0.000 |
| z |
0.000 |
0.000 |
5.395 |
<r2> (average value of r
2) Å
2
| <r2> |
232.338 |
| (<r2>)1/2 |
15.243 |