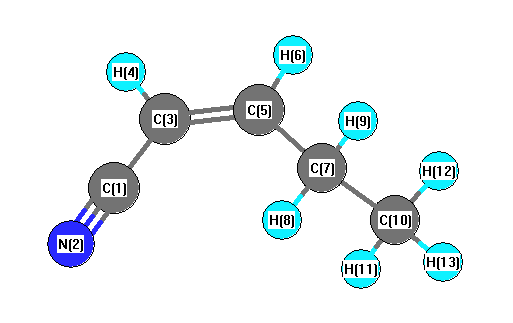
Vibrational Frequencies calculated at wB97X-D/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3221 |
3079 |
2.45 |
|
|
|
| 2 |
A |
3178 |
3038 |
8.38 |
|
|
|
| 3 |
A |
3145 |
3006 |
22.23 |
|
|
|
| 4 |
A |
3137 |
2999 |
24.41 |
|
|
|
| 5 |
A |
3113 |
2976 |
0.38 |
|
|
|
| 6 |
A |
3060 |
2925 |
29.11 |
|
|
|
| 7 |
A |
3057 |
2923 |
10.44 |
|
|
|
| 8 |
A |
2377 |
2272 |
21.44 |
|
|
|
| 9 |
A |
1721 |
1645 |
19.27 |
|
|
|
| 10 |
A |
1510 |
1443 |
8.53 |
|
|
|
| 11 |
A |
1504 |
1438 |
7.33 |
|
|
|
| 12 |
A |
1492 |
1426 |
4.42 |
|
|
|
| 13 |
A |
1436 |
1373 |
0.98 |
|
|
|
| 14 |
A |
1415 |
1353 |
1.57 |
|
|
|
| 15 |
A |
1347 |
1288 |
1.22 |
|
|
|
| 16 |
A |
1304 |
1246 |
0.30 |
|
|
|
| 17 |
A |
1266 |
1211 |
0.71 |
|
|
|
| 18 |
A |
1153 |
1102 |
0.48 |
|
|
|
| 19 |
A |
1102 |
1054 |
3.48 |
|
|
|
| 20 |
A |
1040 |
994 |
3.72 |
|
|
|
| 21 |
A |
1021 |
976 |
0.76 |
|
|
|
| 22 |
A |
968 |
925 |
4.78 |
|
|
|
| 23 |
A |
872 |
833 |
3.43 |
|
|
|
| 24 |
A |
818 |
782 |
6.28 |
|
|
|
| 25 |
A |
771 |
737 |
37.94 |
|
|
|
| 26 |
A |
678 |
648 |
1.59 |
|
|
|
| 27 |
A |
582 |
557 |
1.12 |
|
|
|
| 28 |
A |
404 |
386 |
0.39 |
|
|
|
| 29 |
A |
370 |
354 |
0.06 |
|
|
|
| 30 |
A |
233 |
223 |
1.25 |
|
|
|
| 31 |
A |
207 |
198 |
2.78 |
|
|
|
| 32 |
A |
145 |
139 |
4.72 |
|
|
|
| 33 |
A |
37 |
35 |
1.06 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23841.3 cm
-1
Scaled (by 0.956) Zero Point Vibrational Energy (zpe) 22792.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.059 |
|
|
|
| 2 |
N |
-0.086 |
|
|
|
| 3 |
C |
-0.122 |
|
|
|
| 4 |
H |
0.144 |
|
|
|
| 5 |
C |
-0.110 |
|
|
|
| 6 |
H |
0.137 |
|
|
|
| 7 |
C |
-0.136 |
|
|
|
| 8 |
H |
0.110 |
|
|
|
| 9 |
H |
0.101 |
|
|
|
| 10 |
C |
-0.260 |
|
|
|
| 11 |
H |
0.098 |
|
|
|
| 12 |
H |
0.085 |
|
|
|
| 13 |
H |
0.098 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
3.409 |
2.555 |
0.020 |
4.260 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-42.375 |
-5.631 |
1.175 |
| y |
-5.631 |
-36.032 |
-0.739 |
| z |
1.175 |
-0.739 |
-36.960 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-5.878 |
-5.631 |
1.175 |
| y |
-5.631 |
3.635 |
-0.739 |
| z |
1.175 |
-0.739 |
2.243 |
|
| Polar |
|---|
| 3z2-r2 | 4.486 |
|---|
| x2-y2 | -6.342 |
|---|
| xy | -5.631 |
|---|
| xz | 1.175 |
|---|
| yz | -0.739 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
12.275 |
0.925 |
0.439 |
| y |
0.925 |
8.889 |
-0.236 |
| z |
0.439 |
-0.236 |
6.659 |
<r2> (average value of r
2) Å
2
| <r2> |
197.194 |
| (<r2>)1/2 |
14.043 |