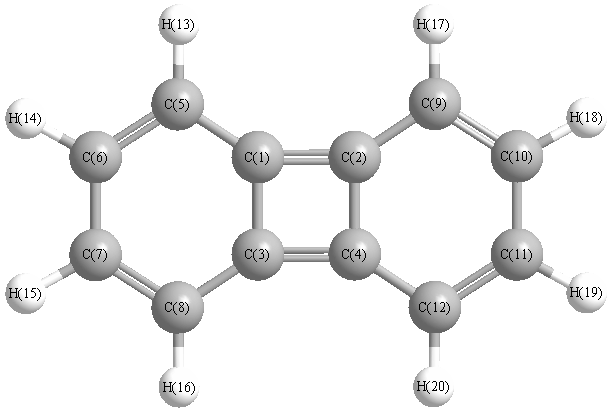
Vibrational Frequencies calculated at wB97X-D/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3226 |
3084 |
0.00 |
|
|
|
| 2 |
Ag |
3210 |
3069 |
0.00 |
|
|
|
| 3 |
Ag |
1741 |
1664 |
0.00 |
|
|
|
| 4 |
Ag |
1529 |
1461 |
0.00 |
|
|
|
| 5 |
Ag |
1446 |
1383 |
0.00 |
|
|
|
| 6 |
Ag |
1206 |
1153 |
0.00 |
|
|
|
| 7 |
Ag |
1146 |
1096 |
0.00 |
|
|
|
| 8 |
Ag |
1021 |
976 |
0.00 |
|
|
|
| 9 |
Ag |
792 |
757 |
0.00 |
|
|
|
| 10 |
Ag |
404 |
386 |
0.00 |
|
|
|
| 11 |
Au |
1006 |
961 |
0.00 |
|
|
|
| 12 |
Au |
912 |
872 |
0.00 |
|
|
|
| 13 |
Au |
831 |
795 |
0.00 |
|
|
|
| 14 |
Au |
601 |
575 |
0.00 |
|
|
|
| 15 |
Au |
146 |
139 |
0.00 |
|
|
|
| 16 |
B1g |
1006 |
962 |
0.00 |
|
|
|
| 17 |
B1g |
893 |
853 |
0.00 |
|
|
|
| 18 |
B1g |
742 |
709 |
0.00 |
|
|
|
| 19 |
B1g |
471 |
450 |
0.00 |
|
|
|
| 20 |
B1u |
3225 |
3084 |
34.12 |
|
|
|
| 21 |
B1u |
3210 |
3069 |
12.89 |
|
|
|
| 22 |
B1u |
1672 |
1598 |
0.07 |
|
|
|
| 23 |
B1u |
1476 |
1411 |
40.26 |
|
|
|
| 24 |
B1u |
1303 |
1246 |
28.81 |
|
|
|
| 25 |
B1u |
1191 |
1139 |
18.74 |
|
|
|
| 26 |
B1u |
1055 |
1009 |
0.03 |
|
|
|
| 27 |
B1u |
999 |
955 |
32.15 |
|
|
|
| 28 |
B1u |
634 |
606 |
3.88 |
|
|
|
| 29 |
B2g |
954 |
912 |
0.00 |
|
|
|
| 30 |
B2g |
766 |
732 |
0.00 |
|
|
|
| 31 |
B2g |
429 |
410 |
0.00 |
|
|
|
| 32 |
B2g |
325 |
310 |
0.00 |
|
|
|
| 33 |
B2u |
3220 |
3078 |
40.48 |
|
|
|
| 34 |
B2u |
3197 |
3056 |
0.41 |
|
|
|
| 35 |
B2u |
1677 |
1603 |
1.76 |
|
|
|
| 36 |
B2u |
1501 |
1435 |
7.44 |
|
|
|
| 37 |
B2u |
1300 |
1243 |
3.64 |
|
|
|
| 38 |
B2u |
1157 |
1107 |
11.98 |
|
|
|
| 39 |
B2u |
1087 |
1039 |
0.35 |
|
|
|
| 40 |
B2u |
741 |
708 |
0.38 |
|
|
|
| 41 |
B2u |
211 |
202 |
0.71 |
|
|
|
| 42 |
B3g |
3219 |
3078 |
0.00 |
|
|
|
| 43 |
B3g |
3197 |
3056 |
0.00 |
|
|
|
| 44 |
B3g |
1681 |
1607 |
0.00 |
|
|
|
| 45 |
B3g |
1503 |
1437 |
0.00 |
|
|
|
| 46 |
B3g |
1318 |
1260 |
0.00 |
|
|
|
| 47 |
B3g |
1123 |
1073 |
0.00 |
|
|
|
| 48 |
B3g |
1014 |
969 |
0.00 |
|
|
|
| 49 |
B3g |
617 |
590 |
0.00 |
|
|
|
| 50 |
B3g |
572 |
547 |
0.00 |
|
|
|
| 51 |
B3u |
948 |
907 |
6.57 |
|
|
|
| 52 |
B3u |
751 |
718 |
139.89 |
|
|
|
| 53 |
B3u |
381 |
365 |
3.82 |
|
|
|
| 54 |
B3u |
107 |
102 |
3.27 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 35043.3 cm
-1
Scaled (by 0.956) Zero Point Vibrational Energy (zpe) 33501.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.017 |
|
|
|
| 2 |
C |
-0.017 |
|
|
|
| 3 |
C |
-0.017 |
|
|
|
| 4 |
C |
-0.017 |
|
|
|
| 5 |
C |
-0.091 |
|
|
|
| 6 |
C |
-0.116 |
|
|
|
| 7 |
C |
-0.116 |
|
|
|
| 8 |
C |
-0.091 |
|
|
|
| 9 |
C |
-0.091 |
|
|
|
| 10 |
C |
-0.116 |
|
|
|
| 11 |
C |
-0.116 |
|
|
|
| 12 |
C |
-0.091 |
|
|
|
| 13 |
H |
0.106 |
|
|
|
| 14 |
H |
0.118 |
|
|
|
| 15 |
H |
0.118 |
|
|
|
| 16 |
H |
0.106 |
|
|
|
| 17 |
H |
0.106 |
|
|
|
| 18 |
H |
0.118 |
|
|
|
| 19 |
H |
0.118 |
|
|
|
| 20 |
H |
0.106 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-74.286 |
0.000 |
0.000 |
| y |
0.000 |
-59.897 |
0.000 |
| z |
0.000 |
0.000 |
-60.014 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-14.330 |
0.000 |
0.000 |
| y |
0.000 |
7.253 |
0.000 |
| z |
0.000 |
0.000 |
7.077 |
|
| Polar |
|---|
| 3z2-r2 | 14.154 |
|---|
| x2-y2 | -14.389 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.818 |
0.000 |
0.000 |
| y |
0.000 |
19.781 |
0.000 |
| z |
0.000 |
0.000 |
31.167 |
<r2> (average value of r
2) Å
2
| <r2> |
556.296 |
| (<r2>)1/2 |
23.586 |