Jump to
S1C2
Energy calculated at wB97X-D/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -345.342495 |
| Energy at 298.15K | |
| HF Energy | -345.342495 |
| Nuclear repulsion energy | 426.586058 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1' |
3069 |
2934 |
0.00 |
|
|
|
| 2 |
A1' |
1509 |
1443 |
0.00 |
|
|
|
| 3 |
A1' |
1366 |
1306 |
0.00 |
|
|
|
| 4 |
A1' |
982 |
939 |
0.00 |
|
|
|
| 5 |
A1' |
834 |
797 |
0.00 |
|
|
|
| 6 |
A1' |
583 |
557 |
0.00 |
|
|
|
| 7 |
A1" |
3092 |
2956 |
0.00 |
|
|
|
| 8 |
A1" |
1265 |
1209 |
0.00 |
|
|
|
| 9 |
A1" |
1025 |
980 |
0.00 |
|
|
|
| 10 |
A1" |
45i |
43i |
0.00 |
|
|
|
| 11 |
A2' |
3114 |
2977 |
0.00 |
|
|
|
| 12 |
A2' |
1199 |
1147 |
0.00 |
|
|
|
| 13 |
A2' |
806 |
770 |
0.00 |
|
|
|
| 14 |
A2" |
3056 |
2922 |
109.08 |
|
|
|
| 15 |
A2" |
1498 |
1432 |
8.19 |
|
|
|
| 16 |
A2" |
1389 |
1328 |
2.89 |
|
|
|
| 17 |
A2" |
1009 |
964 |
22.16 |
|
|
|
| 18 |
A2" |
788 |
753 |
68.98 |
|
|
|
| 19 |
E' |
3120 |
2982 |
89.65 |
|
|
|
| 19 |
E' |
3120 |
2982 |
90.18 |
|
|
|
| 20 |
E' |
3063 |
2928 |
119.05 |
|
|
|
| 20 |
E' |
3063 |
2928 |
119.05 |
|
|
|
| 21 |
E' |
1496 |
1430 |
11.62 |
|
|
|
| 21 |
E' |
1496 |
1430 |
11.69 |
|
|
|
| 22 |
E' |
1367 |
1307 |
13.99 |
|
|
|
| 22 |
E' |
1367 |
1307 |
14.05 |
|
|
|
| 23 |
E' |
1331 |
1273 |
0.32 |
|
|
|
| 23 |
E' |
1331 |
1273 |
0.31 |
|
|
|
| 24 |
E' |
1114 |
1065 |
42.31 |
|
|
|
| 24 |
E' |
1114 |
1065 |
42.26 |
|
|
|
| 25 |
E' |
909 |
869 |
3.39 |
|
|
|
| 25 |
E' |
909 |
869 |
3.42 |
|
|
|
| 26 |
E' |
831 |
794 |
3.97 |
|
|
|
| 26 |
E' |
831 |
794 |
4.00 |
|
|
|
| 27 |
E' |
431 |
412 |
0.02 |
|
|
|
| 27 |
E' |
431 |
412 |
0.01 |
|
|
|
| 28 |
E" |
3094 |
2958 |
0.00 |
|
|
|
| 28 |
E" |
3094 |
2958 |
0.00 |
|
|
|
| 29 |
E" |
3054 |
2920 |
0.00 |
|
|
|
| 29 |
E" |
3054 |
2920 |
0.00 |
|
|
|
| 30 |
E" |
1483 |
1418 |
0.00 |
|
|
|
| 30 |
E" |
1483 |
1418 |
0.00 |
|
|
|
| 31 |
E" |
1358 |
1298 |
0.00 |
|
|
|
| 31 |
E" |
1358 |
1298 |
0.00 |
|
|
|
| 32 |
E" |
1331 |
1272 |
0.00 |
|
|
|
| 32 |
E" |
1331 |
1272 |
0.00 |
|
|
|
| 33 |
E" |
1215 |
1162 |
0.00 |
|
|
|
| 33 |
E" |
1215 |
1162 |
0.00 |
|
|
|
| 34 |
E" |
1064 |
1017 |
0.00 |
|
|
|
| 34 |
E" |
1064 |
1017 |
0.00 |
|
|
|
| 35 |
E" |
594 |
568 |
0.00 |
|
|
|
| 35 |
E" |
594 |
568 |
0.00 |
|
|
|
| 36 |
E" |
332 |
317 |
0.00 |
|
|
|
| 36 |
E" |
332 |
317 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 40454.0 cm
-1
Scaled (by 0.956) Zero Point Vibrational Energy (zpe) 38674.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/cc-pVTZ
Point Group is D3h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.000 |
1.274 |
| N2 |
0.000 |
0.000 |
-1.274 |
| C3 |
0.000 |
1.375 |
0.778 |
| C4 |
1.190 |
-0.687 |
0.778 |
| C5 |
-1.190 |
-0.687 |
0.778 |
| C6 |
0.000 |
1.375 |
-0.778 |
| C7 |
-1.190 |
-0.687 |
-0.778 |
| C8 |
1.190 |
-0.687 |
-0.778 |
| H9 |
0.879 |
1.883 |
1.178 |
| H10 |
-0.879 |
1.883 |
1.178 |
| H11 |
1.191 |
-1.703 |
1.178 |
| H12 |
2.070 |
-0.180 |
1.178 |
| H13 |
-2.070 |
-0.180 |
1.178 |
| H14 |
-1.191 |
-1.703 |
1.178 |
| H15 |
-0.879 |
1.883 |
-1.178 |
| H16 |
0.879 |
1.883 |
-1.178 |
| H17 |
-1.191 |
-1.703 |
-1.178 |
| H18 |
-2.070 |
-0.180 |
-1.178 |
| H19 |
2.070 |
-0.180 |
-1.178 |
| H20 |
1.191 |
-1.703 |
-1.178 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
C3 |
C4 |
C5 |
C6 |
C7 |
C8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
| N1 | | 2.5485 | 1.4614 | 1.4614 | 1.4614 | 2.4703 | 2.4703 | 2.4703 | 2.0800 | 2.0800 | 2.0800 | 2.0800 | 2.0800 | 2.0800 | 3.2139 | 3.2139 | 3.2139 | 3.2139 | 3.2139 | 3.2139 |
N2 | 2.5485 | | 2.4703 | 2.4703 | 2.4703 | 1.4614 | 1.4614 | 1.4614 | 3.2139 | 3.2139 | 3.2139 | 3.2139 | 3.2139 | 3.2139 | 2.0800 | 2.0800 | 2.0800 | 2.0800 | 2.0800 | 2.0800 | C3 | 1.4614 | 2.4703 | | 2.3810 | 2.3810 | 1.5564 | 2.8445 | 2.8445 | 1.0911 | 1.0911 | 3.3238 | 2.6194 | 2.6194 | 3.3238 | 2.2037 | 2.2037 | 3.8358 | 3.2445 | 3.2445 | 3.8358 | C4 | 1.4614 | 2.4703 | 2.3810 | | 2.3810 | 2.8445 | 2.8445 | 1.5564 | 2.6194 | 3.3238 | 1.0911 | 1.0911 | 3.3238 | 2.6194 | 3.8358 | 3.2445 | 3.2445 | 3.8358 | 2.2037 | 2.2037 | C5 | 1.4614 | 2.4703 | 2.3810 | 2.3810 | | 2.8445 | 1.5564 | 2.8445 | 3.3238 | 2.6194 | 2.6194 | 3.3238 | 1.0911 | 1.0911 | 3.2445 | 3.8358 | 2.2037 | 2.2037 | 3.8358 | 3.2445 | C6 | 2.4703 | 1.4614 | 1.5564 | 2.8445 | 2.8445 | | 2.3810 | 2.3810 | 2.2037 | 2.2037 | 3.8358 | 3.2445 | 3.2445 | 3.8358 | 1.0911 | 1.0911 | 3.3238 | 2.6194 | 2.6194 | 3.3238 | C7 | 2.4703 | 1.4614 | 2.8445 | 2.8445 | 1.5564 | 2.3810 | | 2.3810 | 3.8358 | 3.2445 | 3.2445 | 3.8358 | 2.2037 | 2.2037 | 2.6194 | 3.3238 | 1.0911 | 1.0911 | 3.3238 | 2.6194 | C8 | 2.4703 | 1.4614 | 2.8445 | 1.5564 | 2.8445 | 2.3810 | 2.3810 | | 3.2445 | 3.8358 | 2.2037 | 2.2037 | 3.8358 | 3.2445 | 3.3238 | 2.6194 | 2.6194 | 3.3238 | 1.0911 | 1.0911 | H9 | 2.0800 | 3.2139 | 1.0911 | 2.6194 | 3.3238 | 2.2037 | 3.8358 | 3.2445 | | 1.7583 | 3.5988 | 2.3816 | 3.5988 | 4.1400 | 2.9392 | 2.3553 | 4.7631 | 4.3011 | 3.3496 | 4.3011 | H10 | 2.0800 | 3.2139 | 1.0911 | 3.3238 | 2.6194 | 2.2037 | 3.2445 | 3.8358 | 1.7583 | | 4.1400 | 3.5988 | 2.3816 | 3.5988 | 2.3553 | 2.9392 | 4.3011 | 3.3496 | 4.3011 | 4.7631 | H11 | 2.0800 | 3.2139 | 3.3238 | 1.0911 | 2.6194 | 3.8358 | 3.2445 | 2.2037 | 3.5988 | 4.1400 | | 1.7583 | 3.5988 | 2.3816 | 4.7631 | 4.3011 | 3.3496 | 4.3011 | 2.9392 | 2.3553 | H12 | 2.0800 | 3.2139 | 2.6194 | 1.0911 | 3.3238 | 3.2445 | 3.8358 | 2.2037 | 2.3816 | 3.5988 | 1.7583 | | 4.1400 | 3.5988 | 4.3011 | 3.3496 | 4.3011 | 4.7631 | 2.3553 | 2.9392 | H13 | 2.0800 | 3.2139 | 2.6194 | 3.3238 | 1.0911 | 3.2445 | 2.2037 | 3.8358 | 3.5988 | 2.3816 | 3.5988 | 4.1400 | | 1.7583 | 3.3496 | 4.3011 | 2.9392 | 2.3553 | 4.7631 | 4.3011 | H14 | 2.0800 | 3.2139 | 3.3238 | 2.6194 | 1.0911 | 3.8358 | 2.2037 | 3.2445 | 4.1400 | 3.5988 | 2.3816 | 3.5988 | 1.7583 | | 4.3011 | 4.7631 | 2.3553 | 2.9392 | 4.3011 | 3.3496 | H15 | 3.2139 | 2.0800 | 2.2037 | 3.8358 | 3.2445 | 1.0911 | 2.6194 | 3.3238 | 2.9392 | 2.3553 | 4.7631 | 4.3011 | 3.3496 | 4.3011 | | 1.7583 | 3.5988 | 2.3816 | 3.5988 | 4.1400 | H16 | 3.2139 | 2.0800 | 2.2037 | 3.2445 | 3.8358 | 1.0911 | 3.3238 | 2.6194 | 2.3553 | 2.9392 | 4.3011 | 3.3496 | 4.3011 | 4.7631 | 1.7583 | | 4.1400 | 3.5988 | 2.3816 | 3.5988 | H17 | 3.2139 | 2.0800 | 3.8358 | 3.2445 | 2.2037 | 3.3238 | 1.0911 | 2.6194 | 4.7631 | 4.3011 | 3.3496 | 4.3011 | 2.9392 | 2.3553 | 3.5988 | 4.1400 | | 1.7583 | 3.5988 | 2.3816 | H18 | 3.2139 | 2.0800 | 3.2445 | 3.8358 | 2.2037 | 2.6194 | 1.0911 | 3.3238 | 4.3011 | 3.3496 | 4.3011 | 4.7631 | 2.3553 | 2.9392 | 2.3816 | 3.5988 | 1.7583 | | 4.1400 | 3.5988 | H19 | 3.2139 | 2.0800 | 3.2445 | 2.2037 | 3.8358 | 2.6194 | 3.3238 | 1.0911 | 3.3496 | 4.3011 | 2.9392 | 2.3553 | 4.7631 | 4.3011 | 3.5988 | 2.3816 | 3.5988 | 4.1400 | | 1.7583 | H20 | 3.2139 | 2.0800 | 3.8358 | 2.2037 | 3.2445 | 3.3238 | 2.6194 | 1.0911 | 4.3011 | 4.7631 | 2.3553 | 2.9392 | 4.3011 | 3.3496 | 4.1400 | 3.5988 | 2.3816 | 3.5988 | 1.7583 | |
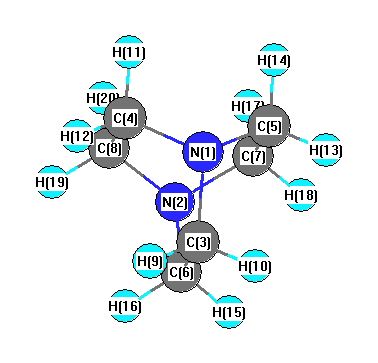 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C3 |
C6 |
109.842 |
|
N1 |
C3 |
H9 |
108.280 |
| N1 |
C3 |
H10 |
108.280 |
|
N1 |
C4 |
C8 |
109.842 |
| N1 |
C4 |
H11 |
108.280 |
|
N1 |
C4 |
H12 |
108.280 |
| N1 |
C5 |
C7 |
109.842 |
|
N1 |
C5 |
H13 |
108.280 |
| N1 |
C5 |
H14 |
108.280 |
|
N2 |
C6 |
C3 |
109.842 |
| N2 |
C6 |
H15 |
108.280 |
|
N2 |
C6 |
H16 |
108.280 |
| N2 |
C7 |
C5 |
109.842 |
|
N2 |
C7 |
H17 |
108.280 |
| N2 |
C7 |
H18 |
108.280 |
|
N2 |
C8 |
C4 |
109.842 |
| N2 |
C8 |
H19 |
108.280 |
|
N2 |
C8 |
H20 |
108.280 |
| C3 |
N1 |
C4 |
109.098 |
|
C3 |
N1 |
C5 |
109.098 |
| C3 |
C6 |
H15 |
111.474 |
|
C3 |
C6 |
H16 |
111.474 |
| C4 |
N1 |
C5 |
109.098 |
|
C4 |
C8 |
H19 |
111.474 |
| C4 |
C8 |
H20 |
111.474 |
|
C5 |
C6 |
H15 |
101.570 |
| C5 |
C6 |
H16 |
151.045 |
|
C6 |
N2 |
C7 |
109.098 |
| C6 |
N2 |
C8 |
109.098 |
|
C6 |
C3 |
H9 |
111.474 |
| C6 |
C3 |
H10 |
111.474 |
|
C7 |
N2 |
C8 |
109.098 |
| C7 |
C5 |
H13 |
111.474 |
|
C7 |
C5 |
H14 |
111.474 |
| C8 |
C4 |
H11 |
111.474 |
|
C8 |
C4 |
H12 |
111.474 |
| H9 |
C3 |
H10 |
107.366 |
|
H11 |
C4 |
H12 |
107.366 |
| H13 |
C5 |
H14 |
107.366 |
|
H15 |
C6 |
H16 |
107.366 |
| H17 |
C7 |
H18 |
107.366 |
|
H19 |
C8 |
H20 |
107.366 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.181 |
|
|
|
| 2 |
N |
-0.181 |
|
|
|
| 3 |
C |
-0.120 |
|
|
|
| 4 |
C |
-0.120 |
|
|
|
| 5 |
C |
-0.120 |
|
|
|
| 6 |
C |
-0.120 |
|
|
|
| 7 |
C |
-0.120 |
|
|
|
| 8 |
C |
-0.120 |
|
|
|
| 9 |
H |
0.090 |
|
|
|
| 10 |
H |
0.090 |
|
|
|
| 11 |
H |
0.090 |
|
|
|
| 12 |
H |
0.090 |
|
|
|
| 13 |
H |
0.090 |
|
|
|
| 14 |
H |
0.090 |
|
|
|
| 15 |
H |
0.090 |
|
|
|
| 16 |
H |
0.090 |
|
|
|
| 17 |
H |
0.090 |
|
|
|
| 18 |
H |
0.090 |
|
|
|
| 19 |
H |
0.090 |
|
|
|
| 20 |
H |
0.090 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.796 |
0.000 |
0.000 |
| y |
0.000 |
-46.796 |
0.000 |
| z |
0.000 |
0.000 |
-57.381 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.292 |
0.000 |
0.000 |
| y |
0.000 |
5.292 |
0.000 |
| z |
0.000 |
0.000 |
-10.585 |
|
| Polar |
|---|
| 3z2-r2 | -21.170 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.892 |
0.000 |
0.000 |
| y |
0.000 |
11.891 |
0.000 |
| z |
0.000 |
0.000 |
10.836 |
<r2> (average value of r
2) Å
2
| <r2> |
212.443 |
| (<r2>)1/2 |
14.575 |
Jump to
S1C1
Energy calculated at wB97X-D/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -345.342580 |
| Energy at 298.15K | -345.359770 |
| HF Energy | -345.342580 |
| Nuclear repulsion energy | 426.587003 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3092 |
2956 |
0.00 |
|
|
|
| 2 |
A1 |
3069 |
2934 |
0.00 |
|
|
|
| 3 |
A1 |
1519 |
1452 |
0.00 |
|
|
|
| 4 |
A1 |
1371 |
1310 |
0.00 |
|
|
|
| 5 |
A1 |
1268 |
1212 |
0.00 |
|
|
|
| 6 |
A1 |
1033 |
988 |
0.00 |
|
|
|
| 7 |
A1 |
984 |
940 |
0.00 |
|
|
|
| 8 |
A1 |
833 |
797 |
0.00 |
|
|
|
| 9 |
A1 |
579 |
553 |
0.00 |
|
|
|
| 10 |
A1 |
52 |
49 |
0.00 |
|
|
|
| 11 |
A2 |
3114 |
2977 |
0.14 |
|
|
|
| 12 |
A2 |
3056 |
2921 |
108.48 |
|
|
|
| 13 |
A2 |
1507 |
1441 |
8.33 |
|
|
|
| 14 |
A2 |
1393 |
1332 |
2.73 |
|
|
|
| 15 |
A2 |
1204 |
1151 |
0.02 |
|
|
|
| 16 |
A2 |
1009 |
964 |
22.12 |
|
|
|
| 17 |
A2 |
817 |
781 |
0.53 |
|
|
|
| 18 |
A2 |
788 |
753 |
67.99 |
|
|
|
| 19 |
E |
3120 |
2982 |
89.05 |
|
|
|
| 19 |
E |
3120 |
2982 |
89.57 |
|
|
|
| 20 |
E |
3094 |
2958 |
0.45 |
|
|
|
| 20 |
E |
3094 |
2958 |
0.45 |
|
|
|
| 21 |
E |
3063 |
2928 |
118.85 |
|
|
|
| 21 |
E |
3063 |
2928 |
118.84 |
|
|
|
| 22 |
E |
3054 |
2920 |
0.13 |
|
|
|
| 22 |
E |
3054 |
2920 |
0.13 |
|
|
|
| 23 |
E |
1504 |
1438 |
11.52 |
|
|
|
| 23 |
E |
1504 |
1438 |
11.60 |
|
|
|
| 24 |
E |
1491 |
1426 |
0.00 |
|
|
|
| 24 |
E |
1491 |
1426 |
0.00 |
|
|
|
| 25 |
E |
1372 |
1312 |
10.82 |
|
|
|
| 25 |
E |
1372 |
1312 |
10.84 |
|
|
|
| 26 |
E |
1360 |
1300 |
2.10 |
|
|
|
| 26 |
E |
1360 |
1300 |
2.07 |
|
|
|
| 27 |
E |
1338 |
1280 |
0.74 |
|
|
|
| 27 |
E |
1338 |
1280 |
0.74 |
|
|
|
| 28 |
E |
1331 |
1273 |
0.01 |
|
|
|
| 28 |
E |
1331 |
1273 |
0.01 |
|
|
|
| 29 |
E |
1218 |
1164 |
0.00 |
|
|
|
| 29 |
E |
1218 |
1164 |
0.00 |
|
|
|
| 30 |
E |
1114 |
1065 |
42.36 |
|
|
|
| 30 |
E |
1114 |
1065 |
42.32 |
|
|
|
| 31 |
E |
1067 |
1020 |
0.00 |
|
|
|
| 31 |
E |
1067 |
1020 |
0.00 |
|
|
|
| 32 |
E |
911 |
871 |
3.50 |
|
|
|
| 32 |
E |
911 |
871 |
3.53 |
|
|
|
| 33 |
E |
841 |
804 |
4.06 |
|
|
|
| 33 |
E |
841 |
804 |
4.04 |
|
|
|
| 34 |
E |
596 |
570 |
0.00 |
|
|
|
| 34 |
E |
596 |
570 |
0.00 |
|
|
|
| 35 |
E |
435 |
416 |
0.01 |
|
|
|
| 35 |
E |
435 |
416 |
0.01 |
|
|
|
| 36 |
E |
340 |
325 |
0.00 |
|
|
|
| 36 |
E |
340 |
325 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 40592.0 cm
-1
Scaled (by 0.956) Zero Point Vibrational Energy (zpe) 38806.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/cc-pVTZ
Point Group is D3
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.000 |
1.279 |
| N2 |
0.000 |
0.000 |
-1.279 |
| C3 |
-0.001 |
1.376 |
0.776 |
| C4 |
1.192 |
-0.687 |
0.776 |
| C5 |
-1.191 |
-0.689 |
0.776 |
| C6 |
0.001 |
1.376 |
-0.776 |
| C7 |
-1.192 |
-0.687 |
-0.776 |
| C8 |
1.191 |
-0.689 |
-0.776 |
| H9 |
0.877 |
1.885 |
1.176 |
| H10 |
-0.882 |
1.882 |
1.174 |
| H11 |
1.194 |
-1.702 |
1.176 |
| H12 |
2.071 |
-0.178 |
1.174 |
| H13 |
-2.071 |
-0.183 |
1.176 |
| H14 |
-1.189 |
-1.705 |
1.174 |
| H15 |
-0.877 |
1.885 |
-1.176 |
| H16 |
0.882 |
1.882 |
-1.174 |
| H17 |
-1.194 |
-1.702 |
-1.176 |
| H18 |
-2.071 |
-0.178 |
-1.174 |
| H19 |
2.071 |
-0.183 |
-1.176 |
| H20 |
1.189 |
-1.705 |
-1.174 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
C3 |
C4 |
C5 |
C6 |
C7 |
C8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
| N1 | | 2.5571 | 1.4647 | 1.4647 | 1.4647 | 2.4728 | 2.4728 | 2.4728 | 2.0820 | 2.0811 | 2.0820 | 2.0811 | 2.0820 | 2.0811 | 3.2167 | 3.2149 | 3.2167 | 3.2149 | 3.2167 | 3.2149 |
N2 | 2.5571 | | 2.4728 | 2.4728 | 2.4728 | 1.4647 | 1.4647 | 1.4647 | 3.2167 | 3.2149 | 3.2167 | 3.2149 | 3.2167 | 3.2149 | 2.0820 | 2.0811 | 2.0820 | 2.0811 | 2.0820 | 2.0811 | C3 | 1.4647 | 2.4728 | | 2.3830 | 2.3830 | 1.5522 | 2.8432 | 2.8447 | 1.0910 | 1.0911 | 3.3261 | 2.6199 | 2.6223 | 3.3256 | 2.1993 | 2.1997 | 3.8352 | 3.2407 | 3.2455 | 3.8352 | C4 | 1.4647 | 2.4728 | 2.3830 | | 2.3830 | 2.8432 | 2.8447 | 1.5522 | 2.6223 | 3.3256 | 1.0910 | 1.0911 | 3.3261 | 2.6199 | 3.8352 | 3.2407 | 3.2455 | 3.8352 | 2.1993 | 2.1997 | C5 | 1.4647 | 2.4728 | 2.3830 | 2.3830 | | 2.8447 | 1.5522 | 2.8432 | 3.3261 | 2.6199 | 2.6223 | 3.3256 | 1.0910 | 1.0911 | 3.2455 | 3.8352 | 2.1993 | 2.1997 | 3.8352 | 3.2407 | C6 | 2.4728 | 1.4647 | 1.5522 | 2.8432 | 2.8447 | | 2.3830 | 2.3830 | 2.1993 | 2.1997 | 3.8352 | 3.2407 | 3.2455 | 3.8352 | 1.0910 | 1.0911 | 3.3261 | 2.6199 | 2.6223 | 3.3256 | C7 | 2.4728 | 1.4647 | 2.8432 | 2.8447 | 1.5522 | 2.3830 | | 2.3830 | 3.8352 | 3.2407 | 3.2455 | 3.8352 | 2.1993 | 2.1997 | 2.6223 | 3.3256 | 1.0910 | 1.0911 | 3.3261 | 2.6199 | C8 | 2.4728 | 1.4647 | 2.8447 | 1.5522 | 2.8432 | 2.3830 | 2.3830 | | 3.2455 | 3.8352 | 2.1993 | 2.1997 | 3.8352 | 3.2407 | 3.3261 | 2.6199 | 2.6223 | 3.3256 | 1.0910 | 1.0911 | H9 | 2.0820 | 3.2167 | 1.0910 | 2.6223 | 3.3261 | 2.1993 | 3.8352 | 3.2455 | | 1.7588 | 3.6017 | 2.3835 | 3.6017 | 4.1423 | 2.9337 | 2.3497 | 4.7635 | 4.2975 | 3.3514 | 4.3019 | H10 | 2.0811 | 3.2149 | 1.0911 | 3.3256 | 2.6199 | 2.1997 | 3.2407 | 3.8352 | 1.7588 | | 4.1423 | 3.6000 | 2.3835 | 3.6000 | 2.3497 | 2.9364 | 4.2975 | 3.3425 | 4.3019 | 4.7611 | H11 | 2.0820 | 3.2167 | 3.3261 | 1.0910 | 2.6223 | 3.8352 | 3.2455 | 2.1993 | 3.6017 | 4.1423 | | 1.7588 | 3.6017 | 2.3835 | 4.7635 | 4.2975 | 3.3514 | 4.3019 | 2.9337 | 2.3497 | H12 | 2.0811 | 3.2149 | 2.6199 | 1.0911 | 3.3256 | 3.2407 | 3.8352 | 2.1997 | 2.3835 | 3.6000 | 1.7588 | | 4.1423 | 3.6000 | 4.2975 | 3.3425 | 4.3019 | 4.7611 | 2.3497 | 2.9364 | H13 | 2.0820 | 3.2167 | 2.6223 | 3.3261 | 1.0910 | 3.2455 | 2.1993 | 3.8352 | 3.6017 | 2.3835 | 3.6017 | 4.1423 | | 1.7588 | 3.3514 | 4.3019 | 2.9337 | 2.3497 | 4.7635 | 4.2975 | H14 | 2.0811 | 3.2149 | 3.3256 | 2.6199 | 1.0911 | 3.8352 | 2.1997 | 3.2407 | 4.1423 | 3.6000 | 2.3835 | 3.6000 | 1.7588 | | 4.3019 | 4.7611 | 2.3497 | 2.9364 | 4.2975 | 3.3425 | H15 | 3.2167 | 2.0820 | 2.1993 | 3.8352 | 3.2455 | 1.0910 | 2.6223 | 3.3261 | 2.9337 | 2.3497 | 4.7635 | 4.2975 | 3.3514 | 4.3019 | | 1.7588 | 3.6017 | 2.3835 | 3.6017 | 4.1423 | H16 | 3.2149 | 2.0811 | 2.1997 | 3.2407 | 3.8352 | 1.0911 | 3.3256 | 2.6199 | 2.3497 | 2.9364 | 4.2975 | 3.3425 | 4.3019 | 4.7611 | 1.7588 | | 4.1423 | 3.6000 | 2.3835 | 3.6000 | H17 | 3.2167 | 2.0820 | 3.8352 | 3.2455 | 2.1993 | 3.3261 | 1.0910 | 2.6223 | 4.7635 | 4.2975 | 3.3514 | 4.3019 | 2.9337 | 2.3497 | 3.6017 | 4.1423 | | 1.7588 | 3.6017 | 2.3835 | H18 | 3.2149 | 2.0811 | 3.2407 | 3.8352 | 2.1997 | 2.6199 | 1.0911 | 3.3256 | 4.2975 | 3.3425 | 4.3019 | 4.7611 | 2.3497 | 2.9364 | 2.3835 | 3.6000 | 1.7588 | | 4.1423 | 3.6000 | H19 | 3.2167 | 2.0820 | 3.2455 | 2.1993 | 3.8352 | 2.6223 | 3.3261 | 1.0910 | 3.3514 | 4.3019 | 2.9337 | 2.3497 | 4.7635 | 4.2975 | 3.6017 | 2.3835 | 3.6017 | 4.1423 | | 1.7588 | H20 | 3.2149 | 2.0811 | 3.8352 | 2.1997 | 3.2407 | 3.3256 | 2.6199 | 1.0911 | 4.3019 | 4.7611 | 2.3497 | 2.9364 | 4.2975 | 3.3425 | 4.1423 | 3.6000 | 2.3835 | 3.6000 | 1.7588 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C3 |
C6 |
110.062 |
|
N1 |
C3 |
H9 |
108.217 |
| N1 |
C3 |
H10 |
108.142 |
|
N1 |
C4 |
C8 |
110.062 |
| N1 |
C4 |
H11 |
108.217 |
|
N1 |
C4 |
H12 |
108.142 |
| N1 |
C5 |
C7 |
110.062 |
|
N1 |
C5 |
H13 |
108.217 |
| N1 |
C5 |
H14 |
108.142 |
|
N2 |
C6 |
C3 |
110.062 |
| N2 |
C6 |
H15 |
108.217 |
|
N2 |
C6 |
H16 |
108.142 |
| N2 |
C7 |
C5 |
110.062 |
|
N2 |
C7 |
H17 |
108.217 |
| N2 |
C7 |
H18 |
108.142 |
|
N2 |
C8 |
C4 |
110.062 |
| N2 |
C8 |
H19 |
108.217 |
|
N2 |
C8 |
H20 |
108.142 |
| C3 |
N1 |
C4 |
108.874 |
|
C3 |
N1 |
C5 |
108.874 |
| C3 |
C6 |
H15 |
111.423 |
|
C3 |
C6 |
H16 |
111.450 |
| C4 |
N1 |
C5 |
108.874 |
|
C4 |
C8 |
H19 |
111.423 |
| C4 |
C8 |
H20 |
111.450 |
|
C5 |
C6 |
H15 |
101.624 |
| C5 |
C6 |
H16 |
150.941 |
|
C6 |
N2 |
C7 |
108.874 |
| C6 |
N2 |
C8 |
108.874 |
|
C6 |
C3 |
H9 |
111.423 |
| C6 |
C3 |
H10 |
111.450 |
|
C7 |
N2 |
C8 |
108.874 |
| C7 |
C5 |
H13 |
111.423 |
|
C7 |
C5 |
H14 |
111.450 |
| C8 |
C4 |
H11 |
111.423 |
|
C8 |
C4 |
H12 |
111.450 |
| H9 |
C3 |
H10 |
107.414 |
|
H11 |
C4 |
H12 |
107.414 |
| H13 |
C5 |
H14 |
107.414 |
|
H15 |
C6 |
H16 |
107.414 |
| H17 |
C7 |
H18 |
107.414 |
|
H19 |
C8 |
H20 |
107.414 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.182 |
|
|
|
| 2 |
N |
-0.182 |
|
|
|
| 3 |
C |
-0.120 |
|
|
|
| 4 |
C |
-0.120 |
|
|
|
| 5 |
C |
-0.120 |
|
|
|
| 6 |
C |
-0.120 |
|
|
|
| 7 |
C |
-0.120 |
|
|
|
| 8 |
C |
-0.120 |
|
|
|
| 9 |
H |
0.089 |
|
|
|
| 10 |
H |
0.091 |
|
|
|
| 11 |
H |
0.089 |
|
|
|
| 12 |
H |
0.091 |
|
|
|
| 13 |
H |
0.089 |
|
|
|
| 14 |
H |
0.091 |
|
|
|
| 15 |
H |
0.089 |
|
|
|
| 16 |
H |
0.091 |
|
|
|
| 17 |
H |
0.089 |
|
|
|
| 18 |
H |
0.091 |
|
|
|
| 19 |
H |
0.089 |
|
|
|
| 20 |
H |
0.091 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.794 |
0.000 |
0.000 |
| y |
0.000 |
-46.794 |
0.000 |
| z |
0.000 |
0.000 |
-57.410 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.308 |
0.000 |
0.000 |
| y |
0.000 |
5.308 |
0.000 |
| z |
0.000 |
0.000 |
-10.616 |
|
| Polar |
|---|
| 3z2-r2 | -21.232 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.891 |
0.000 |
0.001 |
| y |
0.000 |
11.890 |
0.000 |
| z |
0.001 |
0.000 |
10.829 |
<r2> (average value of r
2) Å
2
| <r2> |
212.417 |
| (<r2>)1/2 |
14.575 |