Jump to
S1C2
Energy calculated at wB97X-D/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -232.126004 |
| Energy at 298.15K | -232.129237 |
| HF Energy | -232.126004 |
| Nuclear repulsion energy | 170.695691 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1' |
3060 |
2925 |
0.00 |
|
|
|
| 2 |
A1' |
2434 |
2326 |
0.00 |
|
|
|
| 3 |
A1' |
1425 |
1362 |
0.00 |
|
|
|
| 4 |
A1' |
1286 |
1230 |
0.00 |
|
|
|
| 5 |
A1' |
560 |
536 |
0.00 |
|
|
|
| 6 |
A1" |
69 |
66 |
0.00 |
|
|
|
| 7 |
A2" |
3059 |
2925 |
49.77 |
|
|
|
| 8 |
A2" |
2311 |
2209 |
7.47 |
|
|
|
| 9 |
A2" |
1417 |
1355 |
8.80 |
|
|
|
| 10 |
A2" |
960 |
918 |
2.85 |
|
|
|
| 11 |
E' |
3131 |
2994 |
11.56 |
|
|
|
| 11 |
E' |
3131 |
2994 |
11.47 |
|
|
|
| 12 |
E' |
1485 |
1420 |
17.27 |
|
|
|
| 12 |
E' |
1485 |
1420 |
17.17 |
|
|
|
| 13 |
E' |
1059 |
1013 |
2.11 |
|
|
|
| 13 |
E' |
1059 |
1013 |
2.12 |
|
|
|
| 14 |
E' |
390 |
373 |
4.57 |
|
|
|
| 14 |
E' |
390 |
373 |
4.57 |
|
|
|
| 15 |
E' |
97 |
92 |
7.56 |
|
|
|
| 15 |
E' |
97 |
92 |
7.57 |
|
|
|
| 16 |
E" |
3132 |
2994 |
0.00 |
|
|
|
| 16 |
E" |
3131 |
2994 |
0.00 |
|
|
|
| 17 |
E" |
1485 |
1420 |
0.00 |
|
|
|
| 17 |
E" |
1485 |
1420 |
0.00 |
|
|
|
| 18 |
E" |
1054 |
1007 |
0.00 |
|
|
|
| 18 |
E" |
1054 |
1007 |
0.00 |
|
|
|
| 19 |
E" |
537 |
514 |
0.00 |
|
|
|
| 19 |
E" |
537 |
514 |
0.00 |
|
|
|
| 20 |
E" |
249 |
238 |
0.00 |
|
|
|
| 20 |
E" |
249 |
238 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20910.4 cm
-1
Scaled (by 0.956) Zero Point Vibrational Energy (zpe) 19990.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/cc-pVTZ
Point Group is D3h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.687 |
| C2 |
0.000 |
0.000 |
-0.687 |
| C3 |
0.000 |
0.000 |
1.889 |
| C4 |
0.000 |
0.000 |
-1.889 |
| C5 |
0.000 |
0.000 |
3.344 |
| C6 |
0.000 |
0.000 |
-3.344 |
| H7 |
0.000 |
1.020 |
3.726 |
| H8 |
-0.884 |
-0.510 |
3.726 |
| H9 |
0.884 |
-0.510 |
3.726 |
| H10 |
0.000 |
1.020 |
-3.726 |
| H11 |
0.884 |
-0.510 |
-3.726 |
| H12 |
-0.884 |
-0.510 |
-3.726 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
| C1 | | 1.3747 | 1.2014 | 2.5761 | 2.6565 | 4.0312 | 3.2057 | 3.2057 | 3.2057 | 4.5300 | 4.5300 | 4.5300 |
C2 | 1.3747 | | 2.5761 | 1.2014 | 4.0312 | 2.6565 | 4.5300 | 4.5300 | 4.5300 | 3.2057 | 3.2057 | 3.2057 | C3 | 1.2014 | 2.5761 | | 3.7775 | 1.4551 | 5.2326 | 2.1018 | 2.1018 | 2.1018 | 5.7070 | 5.7070 | 5.7070 | C4 | 2.5761 | 1.2014 | 3.7775 | | 5.2326 | 1.4551 | 5.7070 | 5.7070 | 5.7070 | 2.1018 | 2.1018 | 2.1018 | C5 | 2.6565 | 4.0312 | 1.4551 | 5.2326 | | 6.6877 | 1.0897 | 1.0897 | 1.0897 | 7.1433 | 7.1433 | 7.1433 | C6 | 4.0312 | 2.6565 | 5.2326 | 1.4551 | 6.6877 | | 7.1433 | 7.1433 | 7.1433 | 1.0897 | 1.0897 | 1.0897 | H7 | 3.2057 | 4.5300 | 2.1018 | 5.7070 | 1.0897 | 7.1433 | | 1.7674 | 1.7674 | 7.4525 | 7.6592 | 7.6592 | H8 | 3.2057 | 4.5300 | 2.1018 | 5.7070 | 1.0897 | 7.1433 | 1.7674 | | 1.7674 | 7.6592 | 7.6592 | 7.4525 | H9 | 3.2057 | 4.5300 | 2.1018 | 5.7070 | 1.0897 | 7.1433 | 1.7674 | 1.7674 | | 7.6592 | 7.4525 | 7.6592 | H10 | 4.5300 | 3.2057 | 5.7070 | 2.1018 | 7.1433 | 1.0897 | 7.4525 | 7.6592 | 7.6592 | | 1.7674 | 1.7674 | H11 | 4.5300 | 3.2057 | 5.7070 | 2.1018 | 7.1433 | 1.0897 | 7.6592 | 7.6592 | 7.4525 | 1.7674 | | 1.7674 | H12 | 4.5300 | 3.2057 | 5.7070 | 2.1018 | 7.1433 | 1.0897 | 7.6592 | 7.4525 | 7.6592 | 1.7674 | 1.7674 | |
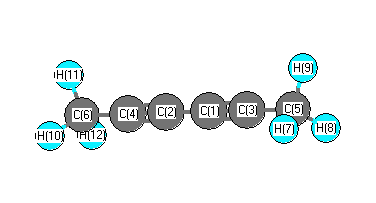 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C4 |
180.000 |
|
C1 |
C3 |
C5 |
180.000 |
| C2 |
C1 |
C3 |
180.000 |
|
C2 |
C4 |
C6 |
180.000 |
| C3 |
C5 |
H7 |
110.544 |
|
C3 |
C5 |
H8 |
110.544 |
| C3 |
C5 |
H9 |
110.544 |
|
C4 |
C6 |
H10 |
110.544 |
| C4 |
C6 |
H11 |
110.544 |
|
C4 |
C6 |
H12 |
110.544 |
| H7 |
C5 |
H8 |
108.378 |
|
H7 |
C5 |
H9 |
108.378 |
| H8 |
C5 |
H9 |
108.378 |
|
H10 |
C6 |
H11 |
108.378 |
| H10 |
C6 |
H12 |
108.378 |
|
H11 |
C6 |
H12 |
108.378 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.475 |
|
|
|
| 2 |
C |
0.475 |
|
|
|
| 3 |
C |
-0.526 |
|
|
|
| 4 |
C |
-0.526 |
|
|
|
| 5 |
C |
-0.261 |
|
|
|
| 6 |
C |
-0.261 |
|
|
|
| 7 |
H |
0.104 |
|
|
|
| 8 |
H |
0.104 |
|
|
|
| 9 |
H |
0.104 |
|
|
|
| 10 |
H |
0.104 |
|
|
|
| 11 |
H |
0.104 |
|
|
|
| 12 |
H |
0.104 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-36.394 |
0.000 |
0.000 |
| y |
0.000 |
-36.394 |
0.000 |
| z |
0.000 |
0.000 |
-19.005 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-8.695 |
0.000 |
0.000 |
| y |
0.000 |
-8.695 |
0.000 |
| z |
0.000 |
0.000 |
17.389 |
|
| Polar |
|---|
| 3z2-r2 | 34.779 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.447 |
0.000 |
0.000 |
| y |
0.000 |
6.446 |
0.000 |
| z |
0.000 |
0.000 |
20.638 |
<r2> (average value of r
2) Å
2
| <r2> |
291.100 |
| (<r2>)1/2 |
17.062 |
Jump to
S1C1
Energy calculated at wB97X-D/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -232.125980 |
| Energy at 298.15K | |
| HF Energy | -232.125980 |
| Nuclear repulsion energy | 170.703856 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3131 |
2994 |
0.00 |
|
|
|
| 2 |
A |
3131 |
2993 |
11.69 |
|
|
|
| 3 |
A |
3129 |
2992 |
0.01 |
|
|
|
| 4 |
A |
3129 |
2992 |
11.62 |
|
|
|
| 5 |
A |
3059 |
2925 |
0.00 |
|
|
|
| 6 |
A |
3059 |
2925 |
50.17 |
|
|
|
| 7 |
A |
2434 |
2327 |
0.00 |
|
|
|
| 8 |
A |
2312 |
2210 |
7.54 |
|
|
|
| 9 |
A |
1486 |
1421 |
17.09 |
|
|
|
| 10 |
A |
1486 |
1420 |
0.02 |
|
|
|
| 11 |
A |
1479 |
1413 |
17.37 |
|
|
|
| 12 |
A |
1478 |
1413 |
0.00 |
|
|
|
| 13 |
A |
1423 |
1360 |
0.04 |
|
|
|
| 14 |
A |
1415 |
1353 |
8.62 |
|
|
|
| 15 |
A |
1286 |
1230 |
0.00 |
|
|
|
| 16 |
A |
1059 |
1012 |
2.27 |
|
|
|
| 17 |
A |
1054 |
1008 |
2.05 |
|
|
|
| 18 |
A |
1053 |
1007 |
0.00 |
|
|
|
| 19 |
A |
1049 |
1002 |
0.00 |
|
|
|
| 20 |
A |
961 |
919 |
2.90 |
|
|
|
| 21 |
A |
560 |
536 |
0.00 |
|
|
|
| 22 |
A |
537 |
514 |
0.00 |
|
|
|
| 23 |
A |
537 |
513 |
0.00 |
|
|
|
| 24 |
A |
390 |
373 |
4.59 |
|
|
|
| 25 |
A |
388 |
371 |
4.50 |
|
|
|
| 26 |
A |
250 |
239 |
0.00 |
|
|
|
| 27 |
A |
246 |
235 |
0.00 |
|
|
|
| 28 |
A |
96 |
92 |
7.53 |
|
|
|
| 29 |
A |
92 |
88 |
7.65 |
|
|
|
| 30 |
A |
55i |
53i |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20827.3 cm
-1
Scaled (by 0.956) Zero Point Vibrational Energy (zpe) 19910.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/cc-pVTZ
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-0.685 |
0.000 |
0.000 |
| C2 |
0.685 |
0.000 |
0.000 |
| C3 |
-1.887 |
0.000 |
0.000 |
| C4 |
1.887 |
0.000 |
0.000 |
| C5 |
-3.339 |
0.000 |
-0.000 |
| C6 |
3.339 |
0.000 |
-0.000 |
| H7 |
-3.728 |
-0.000 |
1.018 |
| H8 |
-3.727 |
0.883 |
-0.510 |
| H9 |
-3.727 |
-0.883 |
-0.510 |
| H10 |
3.728 |
0.000 |
1.018 |
| H11 |
3.727 |
-0.883 |
-0.510 |
| H12 |
3.727 |
0.883 |
-0.510 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
| C1 | | 1.3705 | 1.2019 | 2.5724 | 2.6536 | 4.0240 | 3.2084 | 3.2080 | 3.2080 | 4.5289 | 4.5284 | 4.5284 |
C2 | 1.3705 | | 2.5724 | 1.2019 | 4.0240 | 2.6536 | 4.5289 | 4.5284 | 4.5284 | 3.2084 | 3.2080 | 3.2080 | C3 | 1.2019 | 2.5724 | | 3.7743 | 1.4517 | 5.2260 | 2.1035 | 2.1033 | 2.1033 | 5.7065 | 5.7059 | 5.7059 | C4 | 2.5724 | 1.2019 | 3.7743 | | 5.2260 | 1.4517 | 5.7065 | 5.7059 | 5.7059 | 2.1035 | 2.1033 | 2.1033 | C5 | 2.6536 | 4.0240 | 1.4517 | 5.2260 | | 6.6776 | 1.0906 | 1.0905 | 1.0905 | 7.1396 | 7.1388 | 7.1388 | C6 | 4.0240 | 2.6536 | 5.2260 | 1.4517 | 6.6776 | | 7.1396 | 7.1388 | 7.1388 | 1.0906 | 1.0905 | 1.0905 | H7 | 3.2084 | 4.5289 | 2.1035 | 5.7065 | 1.0906 | 7.1396 | | 1.7647 | 1.7647 | 7.4555 | 7.6607 | 7.6607 | H8 | 3.2080 | 4.5284 | 2.1033 | 5.7059 | 1.0905 | 7.1388 | 1.7647 | | 1.7655 | 7.6607 | 7.6601 | 7.4538 | H9 | 3.2080 | 4.5284 | 2.1033 | 5.7059 | 1.0905 | 7.1388 | 1.7647 | 1.7655 | | 7.6607 | 7.4538 | 7.6601 | H10 | 4.5289 | 3.2084 | 5.7065 | 2.1035 | 7.1396 | 1.0906 | 7.4555 | 7.6607 | 7.6607 | | 1.7647 | 1.7647 | H11 | 4.5284 | 3.2080 | 5.7059 | 2.1033 | 7.1388 | 1.0905 | 7.6607 | 7.6601 | 7.4538 | 1.7647 | | 1.7655 | H12 | 4.5284 | 3.2080 | 5.7059 | 2.1033 | 7.1388 | 1.0905 | 7.6607 | 7.4538 | 7.6601 | 1.7647 | 1.7655 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C4 |
179.994 |
|
C1 |
C3 |
C5 |
179.983 |
| C2 |
C1 |
C3 |
179.994 |
|
C2 |
C4 |
C6 |
179.983 |
| C3 |
C5 |
H7 |
110.872 |
|
C3 |
C5 |
H8 |
110.860 |
| C3 |
C5 |
H9 |
110.860 |
|
C4 |
C6 |
H10 |
110.872 |
| C4 |
C6 |
H11 |
110.860 |
|
C4 |
C6 |
H12 |
110.860 |
| H7 |
C5 |
H8 |
108.020 |
|
H7 |
C5 |
H9 |
108.020 |
| H8 |
C5 |
H9 |
108.090 |
|
H10 |
C6 |
H11 |
108.020 |
| H10 |
C6 |
H12 |
108.020 |
|
H11 |
C6 |
H12 |
108.090 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.473 |
|
|
|
| 2 |
C |
0.473 |
|
|
|
| 3 |
C |
-0.523 |
|
|
|
| 4 |
C |
-0.523 |
|
|
|
| 5 |
C |
-0.262 |
|
|
|
| 6 |
C |
-0.262 |
|
|
|
| 7 |
H |
0.104 |
|
|
|
| 8 |
H |
0.104 |
|
|
|
| 9 |
H |
0.104 |
|
|
|
| 10 |
H |
0.104 |
|
|
|
| 11 |
H |
0.104 |
|
|
|
| 12 |
H |
0.104 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.001 |
0.001 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-18.990 |
0.000 |
0.000 |
| y |
0.000 |
-36.394 |
0.000 |
| z |
0.000 |
0.000 |
-36.401 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
17.407 |
0.000 |
0.000 |
| y |
0.000 |
-8.698 |
0.000 |
| z |
0.000 |
0.000 |
-8.709 |
|
| Polar |
|---|
| 3z2-r2 | -17.418 |
|---|
| x2-y2 | 17.403 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
20.641 |
0.000 |
0.000 |
| y |
0.000 |
6.447 |
0.000 |
| z |
0.000 |
0.000 |
6.445 |
<r2> (average value of r
2) Å
2
| <r2> |
291.095 |
| (<r2>)1/2 |
17.062 |