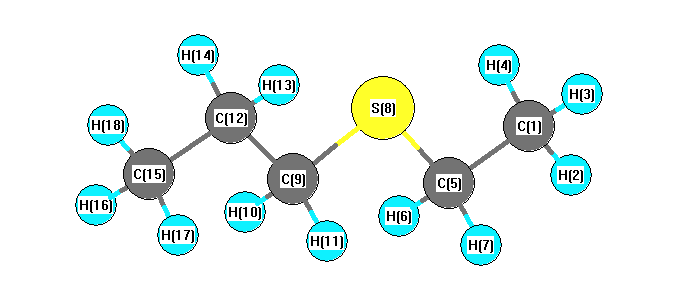
Vibrational Frequencies calculated at wB97X-D/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3135 |
2997 |
22.17 |
|
|
|
| 2 |
A' |
3134 |
2996 |
35.23 |
|
|
|
| 3 |
A' |
3064 |
2930 |
28.46 |
|
|
|
| 4 |
A' |
3061 |
2926 |
30.84 |
|
|
|
| 5 |
A' |
3058 |
2924 |
24.39 |
|
|
|
| 6 |
A' |
3054 |
2920 |
37.74 |
|
|
|
| 7 |
A' |
3051 |
2917 |
14.91 |
|
|
|
| 8 |
A' |
1521 |
1454 |
5.89 |
|
|
|
| 9 |
A' |
1508 |
1441 |
0.88 |
|
|
|
| 10 |
A' |
1506 |
1440 |
3.27 |
|
|
|
| 11 |
A' |
1500 |
1434 |
0.53 |
|
|
|
| 12 |
A' |
1493 |
1427 |
8.02 |
|
|
|
| 13 |
A' |
1432 |
1369 |
2.83 |
|
|
|
| 14 |
A' |
1413 |
1351 |
3.34 |
|
|
|
| 15 |
A' |
1397 |
1336 |
1.23 |
|
|
|
| 16 |
A' |
1312 |
1254 |
9.54 |
|
|
|
| 17 |
A' |
1277 |
1221 |
28.21 |
|
|
|
| 18 |
A' |
1134 |
1084 |
3.10 |
|
|
|
| 19 |
A' |
1073 |
1026 |
3.76 |
|
|
|
| 20 |
A' |
1064 |
1017 |
0.21 |
|
|
|
| 21 |
A' |
1006 |
961 |
4.91 |
|
|
|
| 22 |
A' |
926 |
885 |
2.11 |
|
|
|
| 23 |
A' |
787 |
752 |
1.63 |
|
|
|
| 24 |
A' |
706 |
675 |
0.64 |
|
|
|
| 25 |
A' |
391 |
374 |
0.26 |
|
|
|
| 26 |
A' |
306 |
293 |
0.50 |
|
|
|
| 27 |
A' |
278 |
265 |
0.85 |
|
|
|
| 28 |
A' |
106 |
102 |
0.26 |
|
|
|
| 29 |
A" |
3138 |
3000 |
24.87 |
|
|
|
| 30 |
A" |
3128 |
2991 |
61.05 |
|
|
|
| 31 |
A" |
3108 |
2971 |
2.07 |
|
|
|
| 32 |
A" |
3103 |
2966 |
12.74 |
|
|
|
| 33 |
A" |
3088 |
2952 |
2.05 |
|
|
|
| 34 |
A" |
1502 |
1436 |
7.93 |
|
|
|
| 35 |
A" |
1493 |
1427 |
8.87 |
|
|
|
| 36 |
A" |
1341 |
1282 |
0.19 |
|
|
|
| 37 |
A" |
1270 |
1214 |
0.01 |
|
|
|
| 38 |
A" |
1258 |
1202 |
0.01 |
|
|
|
| 39 |
A" |
1085 |
1037 |
0.86 |
|
|
|
| 40 |
A" |
1047 |
1001 |
0.07 |
|
|
|
| 41 |
A" |
887 |
848 |
0.01 |
|
|
|
| 42 |
A" |
797 |
762 |
4.57 |
|
|
|
| 43 |
A" |
765 |
732 |
2.49 |
|
|
|
| 44 |
A" |
235 |
225 |
0.00 |
|
|
|
| 45 |
A" |
214 |
204 |
0.14 |
|
|
|
| 46 |
A" |
127 |
121 |
1.10 |
|
|
|
| 47 |
A" |
72 |
69 |
0.51 |
|
|
|
| 48 |
A" |
27 |
26 |
0.13 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 35686.8 cm
-1
Scaled (by 0.956) Zero Point Vibrational Energy (zpe) 34116.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.276 |
|
|
|
| 2 |
H |
0.094 |
|
|
|
| 3 |
H |
0.097 |
|
|
|
| 4 |
H |
0.097 |
|
|
|
| 5 |
C |
-0.164 |
|
|
|
| 6 |
H |
0.100 |
|
|
|
| 7 |
H |
0.100 |
|
|
|
| 8 |
S |
-0.102 |
|
|
|
| 9 |
C |
-0.161 |
|
|
|
| 10 |
H |
0.098 |
|
|
|
| 11 |
H |
0.098 |
|
|
|
| 12 |
C |
-0.170 |
|
|
|
| 13 |
H |
0.100 |
|
|
|
| 14 |
H |
0.100 |
|
|
|
| 15 |
C |
-0.277 |
|
|
|
| 16 |
H |
0.085 |
|
|
|
| 17 |
H |
0.085 |
|
|
|
| 18 |
H |
0.096 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.939 |
-1.279 |
0.000 |
1.587 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-43.797 |
2.445 |
0.000 |
| y |
2.445 |
-47.602 |
0.000 |
| z |
0.000 |
0.000 |
-47.716 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.862 |
2.445 |
0.000 |
| y |
2.445 |
-1.846 |
0.000 |
| z |
0.000 |
0.000 |
-2.016 |
|
| Polar |
|---|
| 3z2-r2 | -4.032 |
|---|
| x2-y2 | 3.805 |
|---|
| xy | 2.445 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
13.520 |
2.498 |
0.000 |
| y |
2.498 |
13.298 |
0.000 |
| z |
0.000 |
0.000 |
9.497 |
<r2> (average value of r
2) Å
2
| <r2> |
351.389 |
| (<r2>)1/2 |
18.745 |