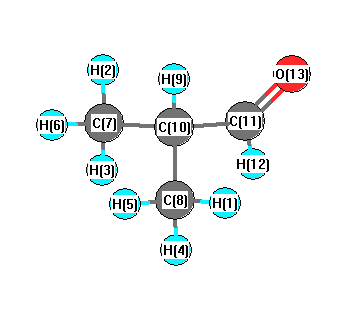
Vibrational Frequencies calculated at wB97X-D/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3137 |
2999 |
27.29 |
|
|
|
| 2 |
A' |
3126 |
2988 |
50.02 |
|
|
|
| 3 |
A' |
3089 |
2953 |
3.83 |
|
|
|
| 4 |
A' |
3050 |
2916 |
18.42 |
|
|
|
| 5 |
A' |
2859 |
2733 |
106.55 |
|
|
|
| 6 |
A' |
1857 |
1776 |
214.06 |
|
|
|
| 7 |
A' |
1517 |
1450 |
17.93 |
|
|
|
| 8 |
A' |
1514 |
1448 |
7.64 |
|
|
|
| 9 |
A' |
1439 |
1376 |
0.71 |
|
|
|
| 10 |
A' |
1423 |
1361 |
4.71 |
|
|
|
| 11 |
A' |
1326 |
1268 |
0.49 |
|
|
|
| 12 |
A' |
1192 |
1140 |
9.34 |
|
|
|
| 13 |
A' |
1190 |
1137 |
0.49 |
|
|
|
| 14 |
A' |
932 |
891 |
18.99 |
|
|
|
| 15 |
A' |
856 |
819 |
21.17 |
|
|
|
| 16 |
A' |
553 |
529 |
5.72 |
|
|
|
| 17 |
A' |
360 |
344 |
7.53 |
|
|
|
| 18 |
A' |
339 |
324 |
0.40 |
|
|
|
| 19 |
A' |
244 |
233 |
0.73 |
|
|
|
| 20 |
A" |
3136 |
2998 |
11.27 |
|
|
|
| 21 |
A" |
3120 |
2982 |
7.18 |
|
|
|
| 22 |
A" |
3049 |
2914 |
23.04 |
|
|
|
| 23 |
A" |
1502 |
1436 |
2.59 |
|
|
|
| 24 |
A" |
1492 |
1426 |
0.26 |
|
|
|
| 25 |
A" |
1408 |
1346 |
4.94 |
|
|
|
| 26 |
A" |
1346 |
1287 |
0.67 |
|
|
|
| 27 |
A" |
1147 |
1097 |
1.07 |
|
|
|
| 28 |
A" |
995 |
951 |
0.21 |
|
|
|
| 29 |
A" |
961 |
918 |
0.40 |
|
|
|
| 30 |
A" |
936 |
895 |
1.43 |
|
|
|
| 31 |
A" |
329 |
314 |
0.64 |
|
|
|
| 32 |
A" |
220 |
210 |
0.02 |
|
|
|
| 33 |
A" |
22 |
21 |
6.38 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 24831.9 cm
-1
Scaled (by 0.956) Zero Point Vibrational Energy (zpe) 23739.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
H |
0.105 |
|
|
|
| 2 |
H |
0.105 |
|
|
|
| 3 |
H |
0.089 |
|
|
|
| 4 |
H |
0.089 |
|
|
|
| 5 |
H |
0.098 |
|
|
|
| 6 |
H |
0.098 |
|
|
|
| 7 |
C |
-0.285 |
|
|
|
| 8 |
C |
-0.285 |
|
|
|
| 9 |
H |
0.101 |
|
|
|
| 10 |
C |
-0.047 |
|
|
|
| 11 |
C |
0.141 |
|
|
|
| 12 |
H |
0.048 |
|
|
|
| 13 |
O |
-0.257 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.464 |
2.837 |
0.000 |
2.875 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.708 |
1.076 |
0.000 |
| y |
1.076 |
-38.267 |
0.000 |
| z |
0.000 |
0.000 |
-30.777 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.814 |
1.076 |
0.000 |
| y |
1.076 |
-7.524 |
0.000 |
| z |
0.000 |
0.000 |
3.711 |
|
| Polar |
|---|
| 3z2-r2 | 7.421 |
|---|
| x2-y2 | 7.559 |
|---|
| xy | 1.076 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.052 |
-0.135 |
0.000 |
| y |
-0.135 |
8.367 |
0.000 |
| z |
0.000 |
0.000 |
7.114 |
<r2> (average value of r
2) Å
2
| <r2> |
133.913 |
| (<r2>)1/2 |
11.572 |