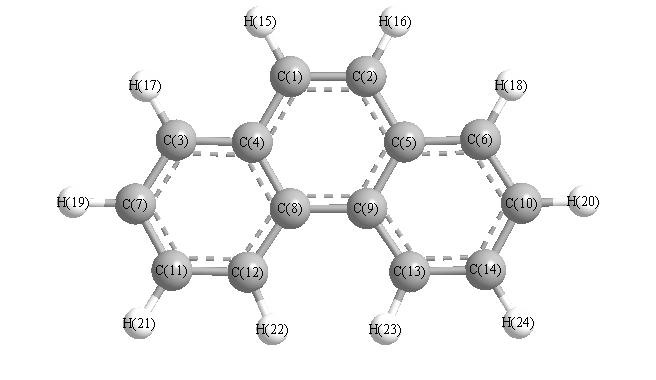
Vibrational Frequencies calculated at wB97X-D/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3242 |
3099 |
17.88 |
|
|
|
| 2 |
A1 |
3222 |
3080 |
0.96 |
|
|
|
| 3 |
A1 |
3209 |
3068 |
28.02 |
|
|
|
| 4 |
A1 |
3204 |
3063 |
15.94 |
|
|
|
| 5 |
A1 |
3195 |
3054 |
1.63 |
|
|
|
| 6 |
A1 |
1708 |
1633 |
0.13 |
|
|
|
| 7 |
A1 |
1681 |
1607 |
2.41 |
|
|
|
| 8 |
A1 |
1592 |
1522 |
2.05 |
|
|
|
| 9 |
A1 |
1495 |
1429 |
1.69 |
|
|
|
| 10 |
A1 |
1480 |
1415 |
4.17 |
|
|
|
| 11 |
A1 |
1396 |
1334 |
0.77 |
|
|
|
| 12 |
A1 |
1338 |
1280 |
2.64 |
|
|
|
| 13 |
A1 |
1288 |
1232 |
10.03 |
|
|
|
| 14 |
A1 |
1227 |
1173 |
1.80 |
|
|
|
| 15 |
A1 |
1206 |
1153 |
0.02 |
|
|
|
| 16 |
A1 |
1176 |
1124 |
0.18 |
|
|
|
| 17 |
A1 |
1129 |
1079 |
1.58 |
|
|
|
| 18 |
A1 |
1076 |
1028 |
1.49 |
|
|
|
| 19 |
A1 |
850 |
813 |
0.14 |
|
|
|
| 20 |
A1 |
734 |
701 |
0.09 |
|
|
|
| 21 |
A1 |
560 |
536 |
0.35 |
|
|
|
| 22 |
A1 |
415 |
397 |
0.51 |
|
|
|
| 23 |
A1 |
249 |
238 |
0.41 |
|
|
|
| 24 |
A2 |
1014 |
969 |
0.00 |
|
|
|
| 25 |
A2 |
1006 |
961 |
0.00 |
|
|
|
| 26 |
A2 |
975 |
932 |
0.00 |
|
|
|
| 27 |
A2 |
901 |
861 |
0.00 |
|
|
|
| 28 |
A2 |
816 |
780 |
0.00 |
|
|
|
| 29 |
A2 |
775 |
741 |
0.00 |
|
|
|
| 30 |
A2 |
611 |
584 |
0.00 |
|
|
|
| 31 |
A2 |
551 |
527 |
0.00 |
|
|
|
| 32 |
A2 |
404 |
386 |
0.00 |
|
|
|
| 33 |
A2 |
242 |
231 |
0.00 |
|
|
|
| 34 |
A2 |
92 |
88 |
0.00 |
|
|
|
| 35 |
B1 |
1014 |
970 |
0.07 |
|
|
|
| 36 |
B1 |
983 |
940 |
3.28 |
|
|
|
| 37 |
B1 |
893 |
854 |
10.72 |
|
|
|
| 38 |
B1 |
840 |
803 |
49.21 |
|
|
|
| 39 |
B1 |
753 |
720 |
83.31 |
|
|
|
| 40 |
B1 |
738 |
706 |
3.12 |
|
|
|
| 41 |
B1 |
512 |
490 |
6.03 |
|
|
|
| 42 |
B1 |
442 |
423 |
8.44 |
|
|
|
| 43 |
B1 |
233 |
223 |
4.13 |
|
|
|
| 44 |
B1 |
94 |
90 |
0.97 |
|
|
|
| 45 |
B2 |
3229 |
3087 |
10.86 |
|
|
|
| 46 |
B2 |
3219 |
3078 |
38.11 |
|
|
|
| 47 |
B2 |
3203 |
3062 |
0.00 |
|
|
|
| 48 |
B2 |
3195 |
3054 |
1.09 |
|
|
|
| 49 |
B2 |
3189 |
3049 |
0.65 |
|
|
|
| 50 |
B2 |
1693 |
1619 |
0.22 |
|
|
|
| 51 |
B2 |
1648 |
1576 |
0.11 |
|
|
|
| 52 |
B2 |
1557 |
1489 |
8.65 |
|
|
|
| 53 |
B2 |
1512 |
1445 |
12.25 |
|
|
|
| 54 |
B2 |
1461 |
1397 |
0.61 |
|
|
|
| 55 |
B2 |
1380 |
1319 |
0.01 |
|
|
|
| 56 |
B2 |
1323 |
1265 |
0.03 |
|
|
|
| 57 |
B2 |
1253 |
1198 |
0.70 |
|
|
|
| 58 |
B2 |
1201 |
1148 |
0.40 |
|
|
|
| 59 |
B2 |
1180 |
1128 |
2.41 |
|
|
|
| 60 |
B2 |
1078 |
1030 |
5.80 |
|
|
|
| 61 |
B2 |
1030 |
985 |
2.08 |
|
|
|
| 62 |
B2 |
899 |
860 |
1.85 |
|
|
|
| 63 |
B2 |
732 |
700 |
2.58 |
|
|
|
| 64 |
B2 |
636 |
608 |
5.31 |
|
|
|
| 65 |
B2 |
510 |
488 |
0.85 |
|
|
|
| 66 |
B2 |
449 |
430 |
2.11 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 43068.3 cm
-1
Scaled (by 0.956) Zero Point Vibrational Energy (zpe) 41173.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.141 |
|
|
|
| 2 |
C |
-0.141 |
|
|
|
| 3 |
C |
-0.134 |
|
|
|
| 4 |
C |
0.088 |
|
|
|
| 5 |
C |
0.088 |
|
|
|
| 6 |
C |
-0.134 |
|
|
|
| 7 |
C |
-0.125 |
|
|
|
| 8 |
C |
0.013 |
|
|
|
| 9 |
C |
0.013 |
|
|
|
| 10 |
C |
-0.125 |
|
|
|
| 11 |
C |
-0.142 |
|
|
|
| 12 |
C |
-0.112 |
|
|
|
| 13 |
C |
-0.112 |
|
|
|
| 14 |
C |
-0.142 |
|
|
|
| 15 |
H |
0.105 |
|
|
|
| 16 |
H |
0.105 |
|
|
|
| 17 |
H |
0.106 |
|
|
|
| 18 |
H |
0.106 |
|
|
|
| 19 |
H |
0.115 |
|
|
|
| 20 |
H |
0.115 |
|
|
|
| 21 |
H |
0.111 |
|
|
|
| 22 |
H |
0.116 |
|
|
|
| 23 |
H |
0.116 |
|
|
|
| 24 |
H |
0.111 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.018 |
0.018 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-87.212 |
0.000 |
0.000 |
| y |
0.000 |
-70.119 |
0.000 |
| z |
0.000 |
0.000 |
-70.464 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-16.920 |
0.000 |
0.000 |
| y |
0.000 |
8.719 |
0.000 |
| z |
0.000 |
0.000 |
8.202 |
|
| Polar |
|---|
| 3z2-r2 | 16.403 |
|---|
| x2-y2 | -17.093 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.118 |
0.000 |
0.000 |
| y |
0.000 |
35.430 |
0.000 |
| z |
0.000 |
0.000 |
24.590 |
<r2> (average value of r
2) Å
2
| <r2> |
726.178 |
| (<r2>)1/2 |
26.948 |