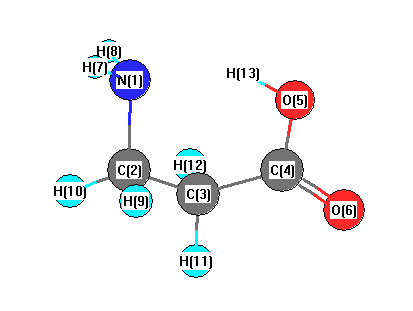
Vibrational Frequencies calculated at wB97X-D/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3586 |
3398 |
11.85 |
|
|
|
| 2 |
A |
3487 |
3305 |
4.53 |
|
|
|
| 3 |
A |
3184 |
3018 |
3.52 |
|
|
|
| 4 |
A |
3158 |
2993 |
12.68 |
|
|
|
| 5 |
A |
3094 |
2932 |
8.78 |
|
|
|
| 6 |
A |
3089 |
2927 |
26.53 |
|
|
|
| 7 |
A |
2593 |
2458 |
1020.09 |
|
|
|
| 8 |
A |
1853 |
1756 |
426.57 |
|
|
|
| 9 |
A |
1718 |
1628 |
48.26 |
|
|
|
| 10 |
A |
1601 |
1517 |
236.45 |
|
|
|
| 11 |
A |
1568 |
1486 |
30.50 |
|
|
|
| 12 |
A |
1527 |
1448 |
12.06 |
|
|
|
| 13 |
A |
1444 |
1369 |
10.24 |
|
|
|
| 14 |
A |
1397 |
1324 |
8.16 |
|
|
|
| 15 |
A |
1350 |
1280 |
4.78 |
|
|
|
| 16 |
A |
1323 |
1254 |
20.49 |
|
|
|
| 17 |
A |
1255 |
1189 |
120.76 |
|
|
|
| 18 |
A |
1228 |
1164 |
116.92 |
|
|
|
| 19 |
A |
1185 |
1123 |
31.58 |
|
|
|
| 20 |
A |
1084 |
1027 |
7.61 |
|
|
|
| 21 |
A |
1029 |
975 |
22.31 |
|
|
|
| 22 |
A |
974 |
924 |
8.20 |
|
|
|
| 23 |
A |
931 |
882 |
42.04 |
|
|
|
| 24 |
A |
871 |
825 |
60.81 |
|
|
|
| 25 |
A |
811 |
768 |
15.17 |
|
|
|
| 26 |
A |
707 |
670 |
13.53 |
|
|
|
| 27 |
A |
582 |
552 |
4.18 |
|
|
|
| 28 |
A |
502 |
476 |
11.54 |
|
|
|
| 29 |
A |
437 |
414 |
10.70 |
|
|
|
| 30 |
A |
371 |
351 |
11.33 |
|
|
|
| 31 |
A |
346 |
328 |
11.25 |
|
|
|
| 32 |
A |
217 |
206 |
5.37 |
|
|
|
| 33 |
A |
91 |
87 |
0.37 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 24295.2 cm
-1
Scaled (by 0.9478) Zero Point Vibrational Energy (zpe) 23026.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.743 |
|
|
|
| 2 |
C |
-0.296 |
|
|
|
| 3 |
C |
-0.554 |
|
|
|
| 4 |
C |
0.688 |
|
|
|
| 5 |
O |
-0.627 |
|
|
|
| 6 |
O |
-0.508 |
|
|
|
| 7 |
H |
0.325 |
|
|
|
| 8 |
H |
0.319 |
|
|
|
| 9 |
H |
0.227 |
|
|
|
| 10 |
H |
0.254 |
|
|
|
| 11 |
H |
0.258 |
|
|
|
| 12 |
H |
0.242 |
|
|
|
| 13 |
H |
0.414 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
7.340 |
-1.599 |
0.626 |
7.538 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-37.699 |
2.654 |
-0.271 |
| y |
2.654 |
-37.143 |
0.258 |
| z |
-0.271 |
0.258 |
-32.741 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.757 |
2.654 |
-0.271 |
| y |
2.654 |
-1.923 |
0.258 |
| z |
-0.271 |
0.258 |
4.680 |
|
| Polar |
|---|
| 3z2-r2 | 9.361 |
|---|
| x2-y2 | -0.556 |
|---|
| xy | 2.654 |
|---|
| xz | -0.271 |
|---|
| yz | 0.258 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.130 |
0.574 |
0.130 |
| y |
0.574 |
5.560 |
-0.051 |
| z |
0.130 |
-0.051 |
4.502 |
<r2> (average value of r
2) Å
2
| <r2> |
169.395 |
| (<r2>)1/2 |
13.015 |