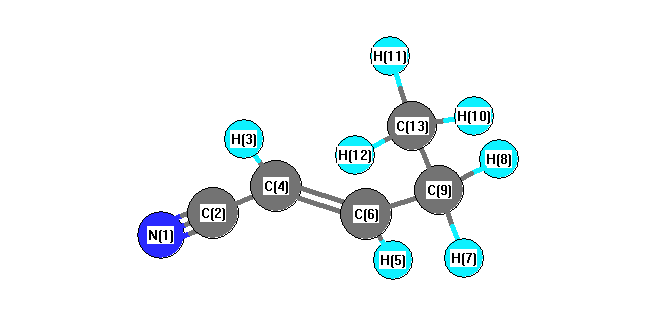
Vibrational Frequencies calculated at wB97X-D/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3234 |
3065 |
1.62 |
|
|
|
| 2 |
A' |
3205 |
3038 |
4.54 |
|
|
|
| 3 |
A' |
3170 |
3005 |
17.54 |
|
|
|
| 4 |
A' |
3087 |
2925 |
11.63 |
|
|
|
| 5 |
A' |
3066 |
2906 |
8.69 |
|
|
|
| 6 |
A' |
2380 |
2255 |
16.01 |
|
|
|
| 7 |
A' |
1741 |
1650 |
20.30 |
|
|
|
| 8 |
A' |
1576 |
1493 |
9.40 |
|
|
|
| 9 |
A' |
1531 |
1451 |
17.92 |
|
|
|
| 10 |
A' |
1476 |
1399 |
14.55 |
|
|
|
| 11 |
A' |
1423 |
1349 |
0.92 |
|
|
|
| 12 |
A' |
1388 |
1315 |
1.18 |
|
|
|
| 13 |
A' |
1348 |
1278 |
8.17 |
|
|
|
| 14 |
A' |
1138 |
1078 |
1.29 |
|
|
|
| 15 |
A' |
1091 |
1034 |
2.47 |
|
|
|
| 16 |
A' |
1014 |
961 |
6.16 |
|
|
|
| 17 |
A' |
904 |
857 |
4.70 |
|
|
|
| 18 |
A' |
606 |
574 |
0.39 |
|
|
|
| 19 |
A' |
550 |
521 |
0.32 |
|
|
|
| 20 |
A' |
284 |
269 |
1.10 |
|
|
|
| 21 |
A' |
163 |
155 |
2.92 |
|
|
|
| 22 |
A" |
3159 |
2994 |
19.08 |
|
|
|
| 23 |
A" |
3096 |
2934 |
4.61 |
|
|
|
| 24 |
A" |
1573 |
1491 |
12.23 |
|
|
|
| 25 |
A" |
1347 |
1277 |
0.00 |
|
|
|
| 26 |
A" |
1153 |
1093 |
0.95 |
|
|
|
| 27 |
A" |
1020 |
966 |
47.38 |
|
|
|
| 28 |
A" |
912 |
865 |
14.68 |
|
|
|
| 29 |
A" |
785 |
744 |
2.54 |
|
|
|
| 30 |
A" |
558 |
529 |
4.02 |
|
|
|
| 31 |
A" |
284 |
269 |
0.05 |
|
|
|
| 32 |
A" |
183 |
173 |
0.05 |
|
|
|
| 33 |
A" |
122 |
116 |
2.56 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 24283.3 cm
-1
Scaled (by 0.9478) Zero Point Vibrational Energy (zpe) 23015.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.484 |
|
|
|
| 2 |
C |
0.289 |
|
|
|
| 3 |
H |
0.264 |
|
|
|
| 4 |
C |
-0.221 |
|
|
|
| 5 |
H |
0.244 |
|
|
|
| 6 |
C |
-0.131 |
|
|
|
| 7 |
H |
0.242 |
|
|
|
| 8 |
H |
0.242 |
|
|
|
| 9 |
C |
-0.483 |
|
|
|
| 10 |
H |
0.224 |
|
|
|
| 11 |
H |
0.219 |
|
|
|
| 12 |
H |
0.219 |
|
|
|
| 13 |
C |
-0.623 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.981 |
-4.144 |
0.000 |
4.594 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-37.240 |
6.689 |
0.000 |
| y |
6.689 |
-44.394 |
0.000 |
| z |
0.000 |
0.000 |
-37.253 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.584 |
6.689 |
0.000 |
| y |
6.689 |
-7.147 |
0.000 |
| z |
0.000 |
0.000 |
3.564 |
|
| Polar |
|---|
| 3z2-r2 | 7.127 |
|---|
| x2-y2 | 7.154 |
|---|
| xy | 6.689 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.088 |
-1.514 |
0.000 |
| y |
-1.514 |
11.332 |
0.000 |
| z |
0.000 |
0.000 |
4.449 |
<r2> (average value of r
2) Å
2
| <r2> |
224.962 |
| (<r2>)1/2 |
14.999 |