Jump to
S1C2
Energy calculated at wB97X-D/3-21G*
| | hartrees |
|---|
| Energy at 0K | -340.401082 |
| Energy at 298.15K | |
| HF Energy | -340.401082 |
| Nuclear repulsion energy | 241.931857 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3148 |
2983 |
21.42 |
|
|
|
| 2 |
A1 |
1935 |
1834 |
500.68 |
|
|
|
| 3 |
A1 |
1591 |
1508 |
0.17 |
|
|
|
| 4 |
A1 |
1384 |
1311 |
0.44 |
|
|
|
| 5 |
A1 |
1080 |
1024 |
182.65 |
|
|
|
| 6 |
A1 |
951 |
901 |
16.35 |
|
|
|
| 7 |
A1 |
855 |
810 |
3.10 |
|
|
|
| 8 |
A1 |
718 |
681 |
2.78 |
|
|
|
| 9 |
A2 |
3190 |
3024 |
0.00 |
|
|
|
| 10 |
A2 |
1277 |
1211 |
0.00 |
|
|
|
| 11 |
A2 |
1142 |
1082 |
0.00 |
|
|
|
| 12 |
A2 |
108i |
102i |
0.00 |
|
|
|
| 13 |
B1 |
3213 |
3045 |
21.51 |
|
|
|
| 14 |
B1 |
1248 |
1183 |
0.11 |
|
|
|
| 15 |
B1 |
880 |
834 |
0.11 |
|
|
|
| 16 |
B1 |
740 |
702 |
29.67 |
|
|
|
| 17 |
B1 |
167 |
158 |
4.45 |
|
|
|
| 18 |
B2 |
3141 |
2977 |
17.41 |
|
|
|
| 19 |
B2 |
1575 |
1493 |
4.98 |
|
|
|
| 20 |
B2 |
1421 |
1347 |
2.87 |
|
|
|
| 21 |
B2 |
1098 |
1041 |
227.87 |
|
|
|
| 22 |
B2 |
1035 |
981 |
77.73 |
|
|
|
| 23 |
B2 |
742 |
703 |
1.46 |
|
|
|
| 24 |
B2 |
514 |
487 |
0.13 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 16467.7 cm
-1
Scaled (by 0.9478) Zero Point Vibrational Energy (zpe) 15608.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/3-21G*
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.891 |
| O2 |
0.000 |
0.000 |
2.084 |
| O3 |
0.000 |
1.120 |
0.071 |
| O4 |
0.000 |
-1.120 |
0.071 |
| C5 |
0.000 |
0.784 |
-1.324 |
| C6 |
0.000 |
-0.784 |
-1.324 |
| H7 |
-0.888 |
1.187 |
-1.817 |
| H8 |
0.888 |
1.187 |
-1.817 |
| H9 |
0.888 |
-1.187 |
-1.817 |
| H10 |
-0.888 |
-1.187 |
-1.817 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.1930 | 1.3880 | 1.3880 | 2.3498 | 2.3498 | 3.0870 | 3.0870 | 3.0870 | 3.0870 |
O2 | 1.1930 | | 2.3035 | 2.3035 | 3.4972 | 3.4972 | 4.1730 | 4.1730 | 4.1730 | 4.1730 | O3 | 1.3880 | 2.3035 | | 2.2397 | 1.4351 | 2.3601 | 2.0873 | 2.0873 | 3.1103 | 3.1103 | O4 | 1.3880 | 2.3035 | 2.2397 | | 2.3601 | 1.4351 | 3.1103 | 3.1103 | 2.0873 | 2.0873 | C5 | 2.3498 | 3.4972 | 1.4351 | 2.3601 | | 1.5675 | 1.0925 | 1.0925 | 2.2170 | 2.2170 | C6 | 2.3498 | 3.4972 | 2.3601 | 1.4351 | 1.5675 | | 2.2170 | 2.2170 | 1.0925 | 1.0925 | H7 | 3.0870 | 4.1730 | 2.0873 | 3.1103 | 1.0925 | 2.2170 | | 1.7757 | 2.9648 | 2.3742 | H8 | 3.0870 | 4.1730 | 2.0873 | 3.1103 | 1.0925 | 2.2170 | 1.7757 | | 2.3742 | 2.9648 | H9 | 3.0870 | 4.1730 | 3.1103 | 2.0873 | 2.2170 | 1.0925 | 2.9648 | 2.3742 | | 1.7757 | H10 | 3.0870 | 4.1730 | 3.1103 | 2.0873 | 2.2170 | 1.0925 | 2.3742 | 2.9648 | 1.7757 | |
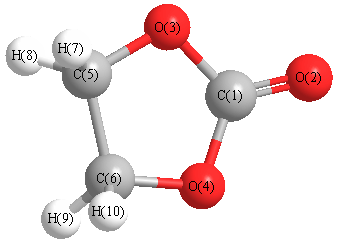 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
112.669 |
|
C1 |
O4 |
C6 |
112.669 |
| O2 |
C1 |
O3 |
126.213 |
|
O2 |
C1 |
O4 |
126.213 |
| O3 |
C1 |
O4 |
107.574 |
|
O3 |
C5 |
C6 |
103.544 |
| O3 |
C5 |
H7 |
110.604 |
|
O3 |
C5 |
H8 |
110.604 |
| O4 |
C6 |
C5 |
103.544 |
|
O4 |
C6 |
H9 |
110.604 |
| O4 |
C6 |
H10 |
110.604 |
|
C5 |
C6 |
H9 |
111.667 |
| C5 |
C6 |
H10 |
111.667 |
|
C6 |
C5 |
H7 |
111.667 |
| C6 |
C5 |
H8 |
111.667 |
|
H7 |
C5 |
H8 |
108.708 |
| H9 |
C6 |
H10 |
108.708 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.919 |
|
|
|
| 2 |
O |
-0.485 |
|
|
|
| 3 |
O |
-0.513 |
|
|
|
| 4 |
O |
-0.513 |
|
|
|
| 5 |
C |
-0.217 |
|
|
|
| 6 |
C |
-0.217 |
|
|
|
| 7 |
H |
0.257 |
|
|
|
| 8 |
H |
0.257 |
|
|
|
| 9 |
H |
0.257 |
|
|
|
| 10 |
H |
0.257 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-5.662 |
5.662 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.640 |
0.000 |
0.000 |
| y |
0.000 |
-36.814 |
0.000 |
| z |
0.000 |
0.000 |
-34.794 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.164 |
0.000 |
0.000 |
| y |
0.000 |
-3.596 |
0.000 |
| z |
0.000 |
0.000 |
-0.567 |
|
| Polar |
|---|
| 3z2-r2 | -1.135 |
|---|
| x2-y2 | 5.173 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.530 |
0.000 |
0.000 |
| y |
0.000 |
4.425 |
0.000 |
| z |
0.000 |
0.000 |
6.848 |
<r2> (average value of r
2) Å
2
| <r2> |
131.795 |
| (<r2>)1/2 |
11.480 |
Jump to
S1C1
Energy calculated at wB97X-D/3-21G*
| | hartrees |
|---|
| Energy at 0K | -340.401822 |
| Energy at 298.15K | -340.408260 |
| HF Energy | -340.401822 |
| Nuclear repulsion energy | 242.526821 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3198 |
3032 |
2.62 |
|
|
|
| 2 |
A |
3133 |
2969 |
13.84 |
|
|
|
| 3 |
A |
1941 |
1839 |
484.67 |
|
|
|
| 4 |
A |
1580 |
1497 |
0.17 |
|
|
|
| 5 |
A |
1394 |
1321 |
1.08 |
|
|
|
| 6 |
A |
1274 |
1207 |
14.76 |
|
|
|
| 7 |
A |
1150 |
1090 |
0.00 |
|
|
|
| 8 |
A |
1069 |
1013 |
164.14 |
|
|
|
| 9 |
A |
957 |
907 |
15.38 |
|
|
|
| 10 |
A |
857 |
812 |
5.20 |
|
|
|
| 11 |
A |
715 |
678 |
2.95 |
|
|
|
| 12 |
A |
169 |
160 |
0.64 |
|
|
|
| 13 |
B |
3213 |
3046 |
15.26 |
|
|
|
| 14 |
B |
3133 |
2969 |
24.00 |
|
|
|
| 15 |
B |
1570 |
1488 |
6.60 |
|
|
|
| 16 |
B |
1418 |
1344 |
1.98 |
|
|
|
| 17 |
B |
1245 |
1180 |
0.73 |
|
|
|
| 18 |
B |
1086 |
1029 |
226.68 |
|
|
|
| 19 |
B |
1034 |
980 |
61.43 |
|
|
|
| 20 |
B |
917 |
869 |
0.65 |
|
|
|
| 21 |
B |
750 |
711 |
30.51 |
|
|
|
| 22 |
B |
681 |
646 |
2.43 |
|
|
|
| 23 |
B |
516 |
489 |
0.06 |
|
|
|
| 24 |
B |
175 |
166 |
4.18 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 16586.3 cm
-1
Scaled (by 0.9478) Zero Point Vibrational Energy (zpe) 15720.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/3-21G*
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.883 |
| O2 |
0.000 |
0.000 |
2.079 |
| O3 |
0.000 |
1.130 |
0.062 |
| O4 |
0.000 |
-1.130 |
0.062 |
| C5 |
0.228 |
0.736 |
-1.318 |
| C6 |
-0.228 |
-0.736 |
-1.318 |
| H7 |
-0.353 |
1.366 |
-1.990 |
| H8 |
1.292 |
0.815 |
-1.560 |
| H9 |
0.353 |
-1.366 |
-1.990 |
| H10 |
-1.292 |
-0.815 |
-1.560 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.1961 | 1.3968 | 1.3968 | 2.3314 | 2.3314 | 3.1999 | 2.8811 | 3.1999 | 2.8811 |
O2 | 1.1961 | | 2.3119 | 2.3119 | 3.4828 | 3.4828 | 4.3058 | 3.9465 | 4.3058 | 3.9465 | O3 | 1.3968 | 2.3119 | | 2.2607 | 1.4531 | 2.3320 | 2.0949 | 2.0977 | 3.2502 | 2.8435 | O4 | 1.3968 | 2.3119 | 2.2607 | | 2.3320 | 1.4531 | 3.2502 | 2.8435 | 2.0949 | 2.0977 | C5 | 2.3314 | 3.4828 | 1.4531 | 2.3320 | | 1.5404 | 1.0888 | 1.0947 | 2.2098 | 2.1849 | C6 | 2.3314 | 3.4828 | 2.3320 | 1.4531 | 1.5404 | | 2.2098 | 2.1849 | 1.0888 | 1.0947 | H7 | 3.1999 | 4.3058 | 2.0949 | 3.2502 | 1.0888 | 2.2098 | | 1.7877 | 2.8215 | 2.4128 | H8 | 2.8811 | 3.9465 | 2.0977 | 2.8435 | 1.0947 | 2.1849 | 1.7877 | | 2.4128 | 3.0556 | H9 | 3.1999 | 4.3058 | 3.2502 | 2.0949 | 2.2098 | 1.0888 | 2.8215 | 2.4128 | | 1.7877 | H10 | 2.8811 | 3.9465 | 2.8435 | 2.0977 | 2.1849 | 1.0947 | 2.4128 | 3.0556 | 1.7877 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
109.764 |
|
C1 |
O4 |
C6 |
109.764 |
| O2 |
C1 |
O3 |
125.980 |
|
O2 |
C1 |
O4 |
125.980 |
| O3 |
C1 |
O4 |
108.041 |
|
O3 |
C5 |
C6 |
102.303 |
| O3 |
C5 |
H7 |
110.179 |
|
O3 |
C5 |
H8 |
110.049 |
| O4 |
C6 |
C5 |
102.303 |
|
O4 |
C6 |
H9 |
110.179 |
| O4 |
C6 |
H10 |
110.049 |
|
C5 |
C6 |
H9 |
113.266 |
| C5 |
C6 |
H10 |
110.893 |
|
C6 |
C5 |
H7 |
113.266 |
| C6 |
C5 |
H8 |
110.893 |
|
H7 |
C5 |
H8 |
109.918 |
| H9 |
C6 |
H10 |
109.918 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.917 |
|
|
|
| 2 |
O |
-0.484 |
|
|
|
| 3 |
O |
-0.511 |
|
|
|
| 4 |
O |
-0.511 |
|
|
|
| 5 |
C |
-0.219 |
|
|
|
| 6 |
C |
-0.219 |
|
|
|
| 7 |
H |
0.259 |
|
|
|
| 8 |
H |
0.255 |
|
|
|
| 9 |
H |
0.259 |
|
|
|
| 10 |
H |
0.255 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-5.588 |
5.588 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.648 |
0.318 |
0.000 |
| y |
0.318 |
-36.885 |
0.000 |
| z |
0.000 |
0.000 |
-34.637 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.112 |
0.318 |
0.000 |
| y |
0.318 |
-3.742 |
0.000 |
| z |
0.000 |
0.000 |
-0.371 |
|
| Polar |
|---|
| 3z2-r2 | -0.741 |
|---|
| x2-y2 | 5.236 |
|---|
| xy | 0.318 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.606 |
0.115 |
0.000 |
| y |
0.115 |
4.362 |
0.000 |
| z |
0.000 |
0.000 |
6.833 |
<r2> (average value of r
2) Å
2
| <r2> |
130.592 |
| (<r2>)1/2 |
11.428 |