Jump to
S1C2
Energy calculated at PBEPBE/6-31G*
| | hartrees |
|---|
| Energy at 0K | -358.079901 |
| Energy at 298.15K | -358.084737 |
| Nuclear repulsion energy | 230.355404 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3643 |
3591 |
64.95 |
|
|
|
| 2 |
A' |
3562 |
3511 |
42.87 |
|
|
|
| 3 |
A' |
3505 |
3455 |
44.47 |
|
|
|
| 4 |
A' |
1785 |
1760 |
107.54 |
|
|
|
| 5 |
A' |
1763 |
1738 |
349.23 |
|
|
|
| 6 |
A' |
1559 |
1537 |
129.01 |
|
|
|
| 7 |
A' |
1385 |
1366 |
11.21 |
|
|
|
| 8 |
A' |
1291 |
1272 |
41.06 |
|
|
|
| 9 |
A' |
1158 |
1142 |
257.41 |
|
|
|
| 10 |
A' |
1066 |
1051 |
2.37 |
|
|
|
| 11 |
A' |
752 |
741 |
7.90 |
|
|
|
| 12 |
A' |
586 |
578 |
64.74 |
|
|
|
| 13 |
A' |
508 |
501 |
0.50 |
|
|
|
| 14 |
A' |
399 |
394 |
4.79 |
|
|
|
| 15 |
A' |
258 |
255 |
14.48 |
|
|
|
| 16 |
A" |
787 |
775 |
5.72 |
|
|
|
| 17 |
A" |
686 |
676 |
128.10 |
|
|
|
| 18 |
A" |
638 |
629 |
8.98 |
|
|
|
| 19 |
A" |
414 |
408 |
11.85 |
|
|
|
| 20 |
A" |
285 |
281 |
235.63 |
|
|
|
| 21 |
A" |
69 |
68 |
0.74 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13050.4 cm
-1
Scaled (by 0.9857) Zero Point Vibrational Energy (zpe) 12863.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/6-31G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.754 |
0.000 |
| C2 |
-0.057 |
-0.794 |
0.000 |
| O3 |
-1.104 |
-1.433 |
0.000 |
| O4 |
1.043 |
1.398 |
0.000 |
| O5 |
-1.233 |
1.296 |
0.000 |
| N6 |
1.206 |
-1.305 |
0.000 |
| H7 |
1.339 |
-2.313 |
0.000 |
| H8 |
2.004 |
-0.672 |
0.000 |
| H9 |
-1.088 |
2.272 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5489 | 2.4498 | 1.2253 | 1.3473 | 2.3867 | 3.3466 | 2.4595 | 1.8670 |
C2 | 1.5489 | | 1.2270 | 2.4516 | 2.3986 | 1.3623 | 2.0629 | 2.0638 | 3.2341 | O3 | 2.4498 | 1.2270 | | 3.5524 | 2.7321 | 2.3136 | 2.5968 | 3.1994 | 3.7044 | O4 | 1.2253 | 2.4516 | 3.5524 | | 2.2785 | 2.7078 | 3.7222 | 2.2819 | 2.3028 | O5 | 1.3473 | 2.3986 | 2.7321 | 2.2785 | | 3.5664 | 4.4321 | 3.7886 | 0.9861 | N6 | 2.3867 | 1.3623 | 2.3136 | 2.7078 | 3.5664 | | 1.0163 | 1.0183 | 4.2492 | H7 | 3.3466 | 2.0629 | 2.5968 | 3.7222 | 4.4321 | 1.0163 | | 1.7702 | 5.1871 | H8 | 2.4595 | 2.0638 | 3.1994 | 2.2819 | 3.7886 | 1.0183 | 1.7702 | | 4.2688 | H9 | 1.8670 | 3.2341 | 3.7044 | 2.3028 | 0.9861 | 4.2492 | 5.1871 | 4.2688 | |
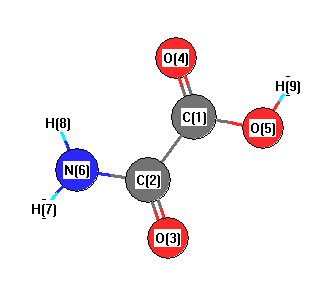 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
123.479 |
|
C1 |
C2 |
N6 |
109.964 |
| C1 |
O5 |
H9 |
105.228 |
|
C2 |
C1 |
O4 |
123.767 |
| C2 |
C1 |
O5 |
111.632 |
|
C2 |
N6 |
H7 |
119.575 |
| C2 |
N6 |
H8 |
119.501 |
|
O3 |
C2 |
N6 |
126.557 |
| O4 |
C1 |
O5 |
124.601 |
|
H7 |
N6 |
H8 |
120.925 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.476 |
|
|
|
| 2 |
C |
0.508 |
|
|
|
| 3 |
O |
-0.448 |
|
|
|
| 4 |
O |
-0.442 |
|
|
|
| 5 |
O |
-0.495 |
|
|
|
| 6 |
N |
-0.718 |
|
|
|
| 7 |
H |
0.350 |
|
|
|
| 8 |
H |
0.361 |
|
|
|
| 9 |
H |
0.408 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.148 |
0.654 |
0.000 |
2.245 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-34.001 |
-9.869 |
0.000 |
| y |
-9.869 |
-27.765 |
0.000 |
| z |
0.000 |
0.000 |
-33.120 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.558 |
-9.869 |
0.000 |
| y |
-9.869 |
5.795 |
0.000 |
| z |
0.000 |
0.000 |
-2.237 |
|
| Polar |
|---|
| 3z2-r2 | -4.473 |
|---|
| x2-y2 | -6.236 |
|---|
| xy | -9.869 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.870 |
-0.274 |
0.000 |
| y |
-0.274 |
6.315 |
0.000 |
| z |
0.000 |
0.000 |
2.507 |
<r2> (average value of r
2) Å
2
| <r2> |
143.140 |
| (<r2>)1/2 |
11.964 |
Jump to
S1C1
Energy calculated at PBEPBE/6-31G*
| | hartrees |
|---|
| Energy at 0K | -358.087473 |
| Energy at 298.15K | -358.092609 |
| Nuclear repulsion energy | 232.079422 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3632 |
3580 |
72.60 |
|
|
|
| 2 |
A' |
3498 |
3448 |
54.65 |
|
|
|
| 3 |
A' |
3297 |
3250 |
139.81 |
|
|
|
| 4 |
A' |
1819 |
1793 |
201.33 |
|
|
|
| 5 |
A' |
1758 |
1733 |
211.79 |
|
|
|
| 6 |
A' |
1564 |
1541 |
72.71 |
|
|
|
| 7 |
A' |
1424 |
1403 |
307.33 |
|
|
|
| 8 |
A' |
1330 |
1311 |
226.41 |
|
|
|
| 9 |
A' |
1187 |
1170 |
7.82 |
|
|
|
| 10 |
A' |
1072 |
1057 |
6.53 |
|
|
|
| 11 |
A' |
780 |
769 |
8.90 |
|
|
|
| 12 |
A' |
609 |
601 |
9.04 |
|
|
|
| 13 |
A' |
528 |
520 |
1.21 |
|
|
|
| 14 |
A' |
394 |
389 |
9.34 |
|
|
|
| 15 |
A' |
267 |
263 |
40.90 |
|
|
|
| 16 |
A" |
803 |
792 |
73.71 |
|
|
|
| 17 |
A" |
760 |
749 |
23.40 |
|
|
|
| 18 |
A" |
663 |
654 |
1.42 |
|
|
|
| 19 |
A" |
444 |
438 |
111.49 |
|
|
|
| 20 |
A" |
347 |
342 |
154.67 |
|
|
|
| 21 |
A" |
124 |
122 |
3.20 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13149.5 cm
-1
Scaled (by 0.9857) Zero Point Vibrational Energy (zpe) 12961.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/6-31G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.015 |
-0.800 |
0.000 |
| C2 |
0.000 |
0.749 |
0.000 |
| O3 |
-1.087 |
1.346 |
0.000 |
| O4 |
1.033 |
-1.470 |
0.000 |
| O5 |
-1.237 |
-1.279 |
0.000 |
| N6 |
1.235 |
1.289 |
0.000 |
| H7 |
1.364 |
2.298 |
0.000 |
| H8 |
2.035 |
0.658 |
0.000 |
| H9 |
-1.802 |
-0.452 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5485 | 2.4123 | 1.2199 | 1.3406 | 2.4192 | 3.3787 | 2.4916 | 1.8492 |
C2 | 1.5485 | | 1.2405 | 2.4480 | 2.3755 | 1.3478 | 2.0641 | 2.0373 | 2.1649 | O3 | 2.4123 | 1.2405 | | 3.5258 | 2.6297 | 2.3224 | 2.6294 | 3.1973 | 1.9348 | O4 | 1.2199 | 2.4480 | 3.5258 | | 2.2790 | 2.7671 | 3.7828 | 2.3524 | 3.0126 | O5 | 1.3406 | 2.3755 | 2.6297 | 2.2790 | | 3.5647 | 4.4229 | 3.8030 | 1.0014 | N6 | 2.4192 | 1.3478 | 2.3224 | 2.7671 | 3.5647 | | 1.0168 | 1.0195 | 3.5000 | H7 | 3.3787 | 2.0641 | 2.6294 | 3.7828 | 4.4229 | 1.0168 | | 1.7718 | 4.1931 | H8 | 2.4916 | 2.0373 | 3.1973 | 2.3524 | 3.8030 | 1.0195 | 1.7718 | | 3.9941 | H9 | 1.8492 | 2.1649 | 1.9348 | 3.0126 | 1.0014 | 3.5000 | 4.1931 | 3.9941 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
119.343 |
|
C1 |
C2 |
N6 |
113.105 |
| C1 |
O5 |
H9 |
103.340 |
|
C2 |
C1 |
O4 |
123.897 |
| C2 |
C1 |
O5 |
110.410 |
|
C2 |
N6 |
H7 |
120.961 |
| C2 |
N6 |
H8 |
118.101 |
|
O3 |
C2 |
N6 |
127.552 |
| O4 |
C1 |
O5 |
125.693 |
|
H7 |
N6 |
H8 |
120.938 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.518 |
|
|
|
| 2 |
C |
0.493 |
|
|
|
| 3 |
O |
-0.490 |
|
|
|
| 4 |
O |
-0.434 |
|
|
|
| 5 |
O |
-0.526 |
|
|
|
| 6 |
N |
-0.714 |
|
|
|
| 7 |
H |
0.357 |
|
|
|
| 8 |
H |
0.371 |
|
|
|
| 9 |
H |
0.424 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.613 |
2.694 |
0.000 |
3.140 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-29.797 |
6.922 |
0.000 |
| y |
6.922 |
-36.276 |
0.000 |
| z |
0.000 |
0.000 |
-33.076 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.879 |
6.922 |
0.000 |
| y |
6.922 |
-4.839 |
0.000 |
| z |
0.000 |
0.000 |
-0.040 |
|
| Polar |
|---|
| 3z2-r2 | -0.079 |
|---|
| x2-y2 | 6.479 |
|---|
| xy | 6.922 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.206 |
0.136 |
0.000 |
| y |
0.136 |
5.794 |
0.000 |
| z |
0.000 |
0.000 |
2.492 |
<r2> (average value of r
2) Å
2
| <r2> |
140.453 |
| (<r2>)1/2 |
11.851 |