Vibrational Frequencies calculated at PBEPBE/6-31G(2df,p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3054 |
3023 |
29.44 |
|
|
|
| 2 |
A' |
2978 |
2948 |
36.18 |
|
|
|
| 3 |
A' |
2972 |
2941 |
17.87 |
|
|
|
| 4 |
A' |
2951 |
2921 |
21.07 |
|
|
|
| 5 |
A' |
2935 |
2905 |
18.31 |
|
|
|
| 6 |
A' |
2762 |
2734 |
173.18 |
|
|
|
| 7 |
A' |
1773 |
1754 |
130.19 |
|
|
|
| 8 |
A' |
1459 |
1444 |
4.78 |
|
|
|
| 9 |
A' |
1442 |
1428 |
0.68 |
|
|
|
| 10 |
A' |
1431 |
1416 |
1.34 |
|
|
|
| 11 |
A' |
1394 |
1380 |
14.50 |
|
|
|
| 12 |
A' |
1372 |
1358 |
9.15 |
|
|
|
| 13 |
A' |
1360 |
1346 |
1.07 |
|
|
|
| 14 |
A' |
1347 |
1333 |
4.99 |
|
|
|
| 15 |
A' |
1313 |
1299 |
20.01 |
|
|
|
| 16 |
A' |
1229 |
1217 |
2.69 |
|
|
|
| 17 |
A' |
1105 |
1093 |
9.82 |
|
|
|
| 18 |
A' |
1056 |
1045 |
0.47 |
|
|
|
| 19 |
A' |
1020 |
1010 |
1.58 |
|
|
|
| 20 |
A' |
899 |
890 |
1.50 |
|
|
|
| 21 |
A' |
868 |
859 |
15.97 |
|
|
|
| 22 |
A' |
673 |
666 |
10.15 |
|
|
|
| 23 |
A' |
383 |
379 |
1.73 |
|
|
|
| 24 |
A' |
278 |
275 |
3.78 |
|
|
|
| 25 |
A' |
129 |
128 |
4.38 |
|
|
|
| 26 |
A" |
3047 |
3015 |
43.27 |
|
|
|
| 27 |
A" |
3021 |
2989 |
23.53 |
|
|
|
| 28 |
A" |
2983 |
2952 |
6.04 |
|
|
|
| 29 |
A" |
2962 |
2931 |
6.57 |
|
|
|
| 30 |
A" |
1447 |
1432 |
5.92 |
|
|
|
| 31 |
A" |
1284 |
1270 |
0.20 |
|
|
|
| 32 |
A" |
1262 |
1249 |
0.05 |
|
|
|
| 33 |
A" |
1191 |
1179 |
0.00 |
|
|
|
| 34 |
A" |
1115 |
1103 |
0.07 |
|
|
|
| 35 |
A" |
942 |
932 |
1.02 |
|
|
|
| 36 |
A" |
823 |
815 |
0.10 |
|
|
|
| 37 |
A" |
720 |
713 |
2.79 |
|
|
|
| 38 |
A" |
639 |
632 |
3.35 |
|
|
|
| 39 |
A" |
230 |
228 |
0.00 |
|
|
|
| 40 |
A" |
190 |
188 |
0.52 |
|
|
|
| 41 |
A" |
109 |
108 |
0.85 |
|
|
|
| 42 |
A" |
61 |
60 |
1.38 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 30103.3 cm
-1
Scaled (by 0.9897) Zero Point Vibrational Energy (zpe) 29793.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
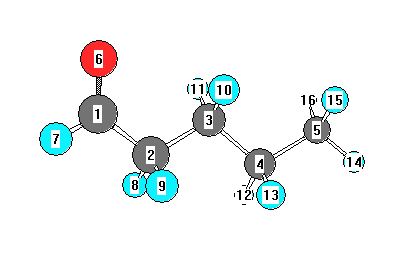
Charges from optimized geometry at PBEPBE/6-31G(2df,p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.124 |
|
|
|
| 2 |
C |
-0.312 |
|
|
|
| 3 |
C |
-0.174 |
|
|
|
| 4 |
C |
-0.198 |
|
|
|
| 5 |
C |
-0.413 |
|
|
|
| 6 |
O |
-0.211 |
|
|
|
| 7 |
H |
0.076 |
|
|
|
| 8 |
H |
0.135 |
|
|
|
| 9 |
H |
0.135 |
|
|
|
| 10 |
H |
0.122 |
|
|
|
| 11 |
H |
0.122 |
|
|
|
| 12 |
H |
0.106 |
|
|
|
| 13 |
H |
0.106 |
|
|
|
| 14 |
H |
0.124 |
|
|
|
| 15 |
H |
0.128 |
|
|
|
| 16 |
H |
0.128 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-2.398 |
0.047 |
0.000 |
2.399 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-43.676 |
3.819 |
0.000 |
| y |
3.819 |
-37.041 |
0.000 |
| z |
0.000 |
0.000 |
-36.845 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-6.733 |
3.819 |
0.000 |
| y |
3.819 |
3.220 |
0.000 |
| z |
0.000 |
0.000 |
3.514 |
|
| Polar |
|---|
| 3z2-r2 | 7.027 |
|---|
| x2-y2 | -6.635 |
|---|
| xy | 3.819 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.057 |
-0.656 |
0.000 |
| y |
-0.656 |
10.446 |
0.000 |
| z |
0.000 |
0.000 |
7.074 |
<r2> (average value of r
2) Å
2
| <r2> |
253.490 |
| (<r2>)1/2 |
15.921 |