Vibrational Frequencies calculated at PBEPBE/6-31G(2df,p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3066 |
3034 |
23.68 |
|
|
|
| 2 |
A |
3062 |
3030 |
25.64 |
|
|
|
| 3 |
A |
3061 |
3029 |
18.01 |
|
|
|
| 4 |
A |
3050 |
3019 |
23.96 |
|
|
|
| 5 |
A |
3047 |
3016 |
41.49 |
|
|
|
| 6 |
A |
3043 |
3011 |
1.12 |
|
|
|
| 7 |
A |
2976 |
2945 |
24.34 |
|
|
|
| 8 |
A |
2975 |
2944 |
34.72 |
|
|
|
| 9 |
A |
2970 |
2939 |
8.09 |
|
|
|
| 10 |
A |
2969 |
2939 |
12.51 |
|
|
|
| 11 |
A |
2947 |
2917 |
3.01 |
|
|
|
| 12 |
A |
2626 |
2599 |
8.09 |
|
|
|
| 13 |
A |
1467 |
1452 |
2.80 |
|
|
|
| 14 |
A |
1455 |
1440 |
2.37 |
|
|
|
| 15 |
A |
1448 |
1433 |
18.58 |
|
|
|
| 16 |
A |
1439 |
1424 |
1.45 |
|
|
|
| 17 |
A |
1435 |
1420 |
4.23 |
|
|
|
| 18 |
A |
1431 |
1417 |
0.47 |
|
|
|
| 19 |
A |
1370 |
1356 |
4.42 |
|
|
|
| 20 |
A |
1357 |
1343 |
8.51 |
|
|
|
| 21 |
A |
1347 |
1333 |
2.54 |
|
|
|
| 22 |
A |
1328 |
1314 |
0.75 |
|
|
|
| 23 |
A |
1296 |
1283 |
0.79 |
|
|
|
| 24 |
A |
1271 |
1258 |
3.52 |
|
|
|
| 25 |
A |
1201 |
1189 |
25.04 |
|
|
|
| 26 |
A |
1162 |
1150 |
2.27 |
|
|
|
| 27 |
A |
1138 |
1126 |
9.24 |
|
|
|
| 28 |
A |
1121 |
1109 |
0.95 |
|
|
|
| 29 |
A |
1057 |
1046 |
1.28 |
|
|
|
| 30 |
A |
1006 |
996 |
10.52 |
|
|
|
| 31 |
A |
953 |
943 |
2.93 |
|
|
|
| 32 |
A |
940 |
931 |
0.74 |
|
|
|
| 33 |
A |
905 |
896 |
1.86 |
|
|
|
| 34 |
A |
899 |
890 |
1.03 |
|
|
|
| 35 |
A |
866 |
857 |
2.53 |
|
|
|
| 36 |
A |
771 |
763 |
2.97 |
|
|
|
| 37 |
A |
672 |
665 |
2.35 |
|
|
|
| 38 |
A |
462 |
457 |
0.41 |
|
|
|
| 39 |
A |
391 |
387 |
0.51 |
|
|
|
| 40 |
A |
360 |
356 |
0.21 |
|
|
|
| 41 |
A |
353 |
349 |
0.21 |
|
|
|
| 42 |
A |
321 |
318 |
1.17 |
|
|
|
| 43 |
A |
241 |
239 |
0.92 |
|
|
|
| 44 |
A |
231 |
229 |
2.84 |
|
|
|
| 45 |
A |
219 |
217 |
0.12 |
|
|
|
| 46 |
A |
192 |
190 |
2.92 |
|
|
|
| 47 |
A |
174 |
172 |
10.93 |
|
|
|
| 48 |
A |
61 |
60 |
1.35 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 34064.8 cm
-1
Scaled (by 0.9897) Zero Point Vibrational Energy (zpe) 33714.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
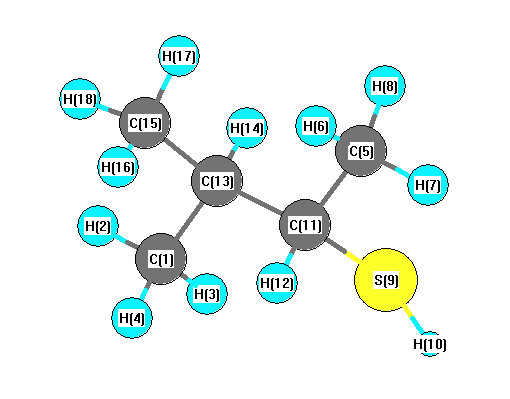
Charges from optimized geometry at PBEPBE/6-31G(2df,p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.431 |
|
|
|
| 2 |
H |
0.123 |
|
|
|
| 3 |
H |
0.154 |
|
|
|
| 4 |
H |
0.126 |
|
|
|
| 5 |
C |
-0.430 |
|
|
|
| 6 |
H |
0.132 |
|
|
|
| 7 |
H |
0.126 |
|
|
|
| 8 |
H |
0.143 |
|
|
|
| 9 |
S |
-0.304 |
|
|
|
| 10 |
H |
0.159 |
|
|
|
| 11 |
C |
0.008 |
|
|
|
| 12 |
H |
0.116 |
|
|
|
| 13 |
C |
0.025 |
|
|
|
| 14 |
H |
0.102 |
|
|
|
| 15 |
C |
-0.431 |
|
|
|
| 16 |
H |
0.125 |
|
|
|
| 17 |
H |
0.129 |
|
|
|
| 18 |
H |
0.128 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.998 |
1.344 |
0.525 |
1.755 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.588 |
2.770 |
1.528 |
| y |
2.770 |
-47.635 |
0.756 |
| z |
1.528 |
0.756 |
-47.719 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.089 |
2.770 |
1.528 |
| y |
2.770 |
-0.481 |
0.756 |
| z |
1.528 |
0.756 |
-0.608 |
|
| Polar |
|---|
| 3z2-r2 | -1.215 |
|---|
| x2-y2 | 1.047 |
|---|
| xy | 2.770 |
|---|
| xz | 1.528 |
|---|
| yz | 0.756 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
12.922 |
-0.246 |
0.033 |
| y |
-0.246 |
11.472 |
0.132 |
| z |
0.033 |
0.132 |
9.125 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |