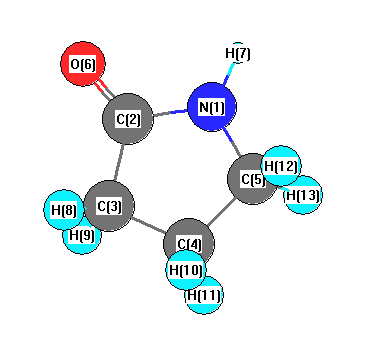
Vibrational Frequencies calculated at PBEPBE/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3493 |
3462 |
18.89 |
|
|
|
| 2 |
A |
3090 |
3062 |
13.23 |
|
|
|
| 3 |
A |
3079 |
3051 |
13.90 |
|
|
|
| 4 |
A |
3030 |
3003 |
39.87 |
|
|
|
| 5 |
A |
3016 |
2989 |
8.89 |
|
|
|
| 6 |
A |
3001 |
2974 |
10.57 |
|
|
|
| 7 |
A |
2945 |
2918 |
59.04 |
|
|
|
| 8 |
A |
1738 |
1722 |
264.95 |
|
|
|
| 9 |
A |
1528 |
1514 |
2.87 |
|
|
|
| 10 |
A |
1497 |
1484 |
7.16 |
|
|
|
| 11 |
A |
1472 |
1459 |
6.72 |
|
|
|
| 12 |
A |
1399 |
1386 |
13.05 |
|
|
|
| 13 |
A |
1337 |
1325 |
2.49 |
|
|
|
| 14 |
A |
1323 |
1311 |
7.83 |
|
|
|
| 15 |
A |
1285 |
1274 |
0.95 |
|
|
|
| 16 |
A |
1246 |
1234 |
15.53 |
|
|
|
| 17 |
A |
1195 |
1184 |
69.49 |
|
|
|
| 18 |
A |
1186 |
1175 |
46.19 |
|
|
|
| 19 |
A |
1168 |
1157 |
2.51 |
|
|
|
| 20 |
A |
1080 |
1070 |
1.59 |
|
|
|
| 21 |
A |
1042 |
1033 |
12.18 |
|
|
|
| 22 |
A |
979 |
970 |
18.12 |
|
|
|
| 23 |
A |
905 |
897 |
0.33 |
|
|
|
| 24 |
A |
893 |
885 |
2.41 |
|
|
|
| 25 |
A |
869 |
862 |
3.55 |
|
|
|
| 26 |
A |
783 |
776 |
8.27 |
|
|
|
| 27 |
A |
668 |
661 |
9.10 |
|
|
|
| 28 |
A |
640 |
635 |
52.06 |
|
|
|
| 29 |
A |
570 |
564 |
51.39 |
|
|
|
| 30 |
A |
497 |
493 |
49.50 |
|
|
|
| 31 |
A |
443 |
439 |
6.27 |
|
|
|
| 32 |
A |
205 |
203 |
2.12 |
|
|
|
| 33 |
A |
150 |
148 |
0.32 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23875.5 cm
-1
Scaled (by 0.9909) Zero Point Vibrational Energy (zpe) 23658.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.639 |
|
|
|
| 2 |
C |
0.600 |
|
|
|
| 3 |
C |
-0.500 |
|
|
|
| 4 |
C |
-0.429 |
|
|
|
| 5 |
C |
-0.227 |
|
|
|
| 6 |
O |
-0.472 |
|
|
|
| 7 |
H |
0.317 |
|
|
|
| 8 |
H |
0.237 |
|
|
|
| 9 |
H |
0.236 |
|
|
|
| 10 |
H |
0.214 |
|
|
|
| 11 |
H |
0.228 |
|
|
|
| 12 |
H |
0.214 |
|
|
|
| 13 |
H |
0.221 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-3.593 |
-0.837 |
0.268 |
3.699 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-42.477 |
-0.212 |
0.244 |
| y |
-0.212 |
-31.148 |
0.130 |
| z |
0.244 |
0.130 |
-35.493 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-9.156 |
-0.212 |
0.244 |
| y |
-0.212 |
7.837 |
0.130 |
| z |
0.244 |
0.130 |
1.319 |
|
| Polar |
|---|
| 3z2-r2 | 2.639 |
|---|
| x2-y2 | -11.329 |
|---|
| xy | -0.212 |
|---|
| xz | 0.244 |
|---|
| yz | 0.130 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.250 |
0.472 |
-0.173 |
| y |
0.472 |
7.055 |
0.082 |
| z |
-0.173 |
0.082 |
5.034 |
<r2> (average value of r
2) Å
2
| <r2> |
149.115 |
| (<r2>)1/2 |
12.211 |