Jump to
S1C2
Energy calculated at PBEPBE/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -271.531485 |
| Energy at 298.15K | |
| HF Energy | -271.531485 |
| Nuclear repulsion energy | 235.000406 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3086 |
3049 |
9.49 |
|
|
|
| 2 |
A' |
3039 |
3002 |
33.06 |
|
|
|
| 3 |
A' |
2983 |
2947 |
18.59 |
|
|
|
| 4 |
A' |
2971 |
2935 |
4.50 |
|
|
|
| 5 |
A' |
2960 |
2925 |
26.68 |
|
|
|
| 6 |
A' |
2937 |
2901 |
15.50 |
|
|
|
| 7 |
A' |
1725 |
1704 |
141.46 |
|
|
|
| 8 |
A' |
1455 |
1437 |
6.78 |
|
|
|
| 9 |
A' |
1437 |
1419 |
2.79 |
|
|
|
| 10 |
A' |
1412 |
1394 |
20.65 |
|
|
|
| 11 |
A' |
1391 |
1375 |
4.53 |
|
|
|
| 12 |
A' |
1358 |
1342 |
1.20 |
|
|
|
| 13 |
A' |
1339 |
1322 |
44.46 |
|
|
|
| 14 |
A' |
1325 |
1309 |
34.38 |
|
|
|
| 15 |
A' |
1272 |
1256 |
21.09 |
|
|
|
| 16 |
A' |
1142 |
1128 |
61.93 |
|
|
|
| 17 |
A' |
1097 |
1083 |
0.46 |
|
|
|
| 18 |
A' |
1034 |
1021 |
0.17 |
|
|
|
| 19 |
A' |
925 |
914 |
3.75 |
|
|
|
| 20 |
A' |
886 |
876 |
13.35 |
|
|
|
| 21 |
A' |
797 |
788 |
3.70 |
|
|
|
| 22 |
A' |
578 |
571 |
8.98 |
|
|
|
| 23 |
A' |
381 |
376 |
1.35 |
|
|
|
| 24 |
A' |
328 |
324 |
2.17 |
|
|
|
| 25 |
A' |
163 |
161 |
4.68 |
|
|
|
| 26 |
A" |
3033 |
2996 |
56.76 |
|
|
|
| 27 |
A" |
3032 |
2995 |
0.40 |
|
|
|
| 28 |
A" |
3007 |
2971 |
0.02 |
|
|
|
| 29 |
A" |
2962 |
2926 |
6.37 |
|
|
|
| 30 |
A" |
1448 |
1430 |
7.12 |
|
|
|
| 31 |
A" |
1421 |
1403 |
11.08 |
|
|
|
| 32 |
A" |
1276 |
1261 |
0.09 |
|
|
|
| 33 |
A" |
1204 |
1189 |
0.02 |
|
|
|
| 34 |
A" |
1096 |
1083 |
0.71 |
|
|
|
| 35 |
A" |
937 |
926 |
0.82 |
|
|
|
| 36 |
A" |
798 |
788 |
0.80 |
|
|
|
| 37 |
A" |
697 |
689 |
4.00 |
|
|
|
| 38 |
A" |
457 |
452 |
0.01 |
|
|
|
| 39 |
A" |
238 |
235 |
0.05 |
|
|
|
| 40 |
A" |
85 |
84 |
0.07 |
|
|
|
| 41 |
A" |
67 |
66 |
0.79 |
|
|
|
| 42 |
A" |
51i |
51i |
0.41 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 29861.2 cm
-1
Scaled (by 0.9879) Zero Point Vibrational Energy (zpe) 29499.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/Def2TZVPP
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-2.416 |
1.363 |
0.000 |
| C2 |
-1.472 |
0.160 |
0.000 |
| C3 |
0.000 |
0.566 |
0.000 |
| C4 |
0.991 |
-0.589 |
0.000 |
| C5 |
2.457 |
-0.201 |
0.000 |
| O6 |
0.629 |
-1.754 |
0.000 |
| H7 |
-3.471 |
1.053 |
0.000 |
| H8 |
-2.261 |
1.998 |
0.886 |
| H9 |
-2.261 |
1.998 |
-0.886 |
| H10 |
-1.669 |
-0.479 |
0.874 |
| H11 |
-1.669 |
-0.479 |
-0.874 |
| H12 |
0.227 |
1.202 |
-0.875 |
| H13 |
0.227 |
1.202 |
0.875 |
| H14 |
3.098 |
-1.090 |
0.000 |
| H15 |
2.694 |
0.412 |
-0.883 |
| H16 |
2.694 |
0.412 |
0.883 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
| C1 | | 1.5289 | 2.5440 | 3.9264 | 5.1179 | 4.3574 | 1.0994 | 1.1007 | 1.1007 | 2.1715 | 2.1715 | 2.7884 | 2.7884 | 6.0353 | 5.2728 | 5.2728 |
C2 | 1.5289 | | 1.5273 | 2.5744 | 3.9460 | 2.8424 | 2.1887 | 2.1869 | 2.1869 | 1.1006 | 1.1006 | 2.1766 | 2.1766 | 4.7385 | 4.2668 | 4.2668 | C3 | 2.5440 | 1.5273 | | 1.5221 | 2.5741 | 2.4038 | 3.5046 | 2.8187 | 2.8187 | 2.1545 | 2.1545 | 1.1049 | 1.1049 | 3.5133 | 2.8398 | 2.8398 | C4 | 3.9264 | 2.5744 | 1.5221 | | 1.5171 | 1.2191 | 4.7539 | 4.2484 | 4.2484 | 2.8016 | 2.8016 | 2.1349 | 2.1349 | 2.1664 | 2.1646 | 2.1646 | C5 | 5.1179 | 3.9460 | 2.5741 | 1.5171 | | 2.3984 | 6.0589 | 5.2800 | 5.2800 | 4.2267 | 4.2267 | 2.7763 | 2.7763 | 1.0962 | 1.1008 | 1.1008 | O6 | 4.3574 | 2.8424 | 2.4038 | 1.2191 | 2.3984 | | 4.9685 | 4.8176 | 4.8176 | 2.7695 | 2.7695 | 3.1087 | 3.1087 | 2.5566 | 3.1199 | 3.1199 | H7 | 1.0994 | 2.1887 | 3.5046 | 4.7539 | 6.0589 | 4.9685 | | 1.7722 | 1.7722 | 2.5214 | 2.5214 | 3.8025 | 3.8025 | 6.9097 | 6.2611 | 6.2611 | H8 | 1.1007 | 2.1869 | 2.8187 | 4.2484 | 5.2800 | 4.8176 | 1.7722 | | 1.7720 | 2.5462 | 3.0954 | 3.1499 | 2.6118 | 6.2482 | 5.4957 | 5.2031 | H9 | 1.1007 | 2.1869 | 2.8187 | 4.2484 | 5.2800 | 4.8176 | 1.7722 | 1.7720 | | 3.0954 | 2.5462 | 2.6118 | 3.1499 | 6.2482 | 5.2031 | 5.4957 | H10 | 2.1715 | 1.1006 | 2.1545 | 2.8016 | 4.2267 | 2.7695 | 2.5214 | 2.5462 | 3.0954 | | 1.7486 | 3.0786 | 2.5336 | 4.8850 | 4.7875 | 4.4532 | H11 | 2.1715 | 1.1006 | 2.1545 | 2.8016 | 4.2267 | 2.7695 | 2.5214 | 3.0954 | 2.5462 | 1.7486 | | 2.5336 | 3.0786 | 4.8850 | 4.4532 | 4.7875 | H12 | 2.7884 | 2.1766 | 1.1049 | 2.1349 | 2.7763 | 3.1087 | 3.8025 | 3.1499 | 2.6118 | 3.0786 | 2.5336 | | 1.7494 | 3.7769 | 2.5913 | 3.1314 | H13 | 2.7884 | 2.1766 | 1.1049 | 2.1349 | 2.7763 | 3.1087 | 3.8025 | 2.6118 | 3.1499 | 2.5336 | 3.0786 | 1.7494 | | 3.7769 | 3.1314 | 2.5913 | H14 | 6.0353 | 4.7385 | 3.5133 | 2.1664 | 1.0962 | 2.5566 | 6.9097 | 6.2482 | 6.2482 | 4.8850 | 4.8850 | 3.7769 | 3.7769 | | 1.7884 | 1.7884 | H15 | 5.2728 | 4.2668 | 2.8398 | 2.1646 | 1.1008 | 3.1199 | 6.2611 | 5.4957 | 5.2031 | 4.7875 | 4.4532 | 2.5913 | 3.1314 | 1.7884 | | 1.7667 | H16 | 5.2728 | 4.2668 | 2.8398 | 2.1646 | 1.1008 | 3.1199 | 6.2611 | 5.2031 | 5.4957 | 4.4532 | 4.7875 | 3.1314 | 2.5913 | 1.7884 | 1.7667 | |
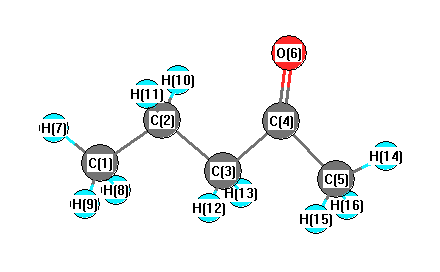 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
112.694 |
|
C1 |
C2 |
H10 |
110.280 |
| C1 |
C2 |
H11 |
110.280 |
|
C2 |
C1 |
H7 |
111.725 |
| C2 |
C1 |
H8 |
111.503 |
|
C2 |
C1 |
H9 |
111.503 |
| C2 |
C3 |
C4 |
115.178 |
|
C2 |
C3 |
H12 |
110.541 |
| C2 |
C3 |
H13 |
110.541 |
|
C3 |
C2 |
H10 |
109.057 |
| C3 |
C2 |
H11 |
109.057 |
|
C3 |
C4 |
C5 |
115.766 |
| C3 |
C4 |
O6 |
122.159 |
|
C4 |
C3 |
H12 |
107.659 |
| C4 |
C3 |
H13 |
107.659 |
|
C4 |
C5 |
H14 |
110.959 |
| C4 |
C5 |
H15 |
110.541 |
|
C4 |
C5 |
H16 |
110.541 |
| C5 |
C4 |
O6 |
122.075 |
|
H7 |
C1 |
H8 |
107.321 |
| H7 |
C1 |
H9 |
107.321 |
|
H8 |
C1 |
H9 |
107.214 |
| H10 |
C2 |
H11 |
105.188 |
|
H12 |
C3 |
H13 |
104.682 |
| H14 |
C5 |
H15 |
108.975 |
|
H14 |
C5 |
H16 |
108.975 |
| H15 |
C5 |
H16 |
106.737 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.252 |
|
|
|
| 2 |
C |
-0.065 |
|
|
|
| 3 |
C |
-0.110 |
|
|
|
| 4 |
C |
0.150 |
|
|
|
| 5 |
C |
-0.232 |
|
|
|
| 6 |
O |
-0.258 |
|
|
|
| 7 |
H |
0.084 |
|
|
|
| 8 |
H |
0.072 |
|
|
|
| 9 |
H |
0.072 |
|
|
|
| 10 |
H |
0.065 |
|
|
|
| 11 |
H |
0.065 |
|
|
|
| 12 |
H |
0.066 |
|
|
|
| 13 |
H |
0.066 |
|
|
|
| 14 |
H |
0.100 |
|
|
|
| 15 |
H |
0.087 |
|
|
|
| 16 |
H |
0.087 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.674 |
2.589 |
0.000 |
2.675 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-35.891 |
1.462 |
0.000 |
| y |
1.462 |
-44.509 |
0.000 |
| z |
0.000 |
0.000 |
-37.517 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.122 |
1.462 |
0.000 |
| y |
1.462 |
-7.805 |
0.000 |
| z |
0.000 |
0.000 |
2.683 |
|
| Polar |
|---|
| 3z2-r2 | 5.367 |
|---|
| x2-y2 | 8.618 |
|---|
| xy | 1.462 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.743 |
-0.550 |
0.000 |
| y |
-0.550 |
10.145 |
0.000 |
| z |
0.000 |
0.000 |
8.005 |
<r2> (average value of r
2) Å
2
| <r2> |
229.806 |
| (<r2>)1/2 |
15.159 |
Jump to
S1C1
Energy calculated at PBEPBE/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -271.531485 |
| Energy at 298.15K | |
| HF Energy | -271.531485 |
| Nuclear repulsion energy | 235.002783 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3086 |
3049 |
9.47 |
|
|
|
| 2 |
A' |
3038 |
3002 |
33.04 |
|
|
|
| 3 |
A' |
2982 |
2946 |
18.62 |
|
|
|
| 4 |
A' |
2970 |
2934 |
4.49 |
|
|
|
| 5 |
A' |
2960 |
2924 |
26.64 |
|
|
|
| 6 |
A' |
2936 |
2900 |
15.53 |
|
|
|
| 7 |
A' |
1725 |
1704 |
141.30 |
|
|
|
| 8 |
A' |
1455 |
1437 |
6.78 |
|
|
|
| 9 |
A' |
1437 |
1419 |
2.79 |
|
|
|
| 10 |
A' |
1412 |
1395 |
20.69 |
|
|
|
| 11 |
A' |
1391 |
1375 |
4.57 |
|
|
|
| 12 |
A' |
1358 |
1342 |
1.19 |
|
|
|
| 13 |
A' |
1339 |
1323 |
44.87 |
|
|
|
| 14 |
A' |
1325 |
1309 |
34.21 |
|
|
|
| 15 |
A' |
1272 |
1257 |
20.92 |
|
|
|
| 16 |
A' |
1142 |
1128 |
61.87 |
|
|
|
| 17 |
A' |
1097 |
1083 |
0.45 |
|
|
|
| 18 |
A' |
1033 |
1021 |
0.16 |
|
|
|
| 19 |
A' |
925 |
914 |
3.73 |
|
|
|
| 20 |
A' |
886 |
876 |
13.40 |
|
|
|
| 21 |
A' |
798 |
788 |
3.65 |
|
|
|
| 22 |
A' |
578 |
571 |
8.99 |
|
|
|
| 23 |
A' |
381 |
376 |
1.35 |
|
|
|
| 24 |
A' |
328 |
324 |
2.17 |
|
|
|
| 25 |
A' |
163 |
161 |
4.68 |
|
|
|
| 26 |
A" |
3033 |
2996 |
57.16 |
|
|
|
| 27 |
A" |
3031 |
2995 |
0.01 |
|
|
|
| 28 |
A" |
3007 |
2971 |
0.00 |
|
|
|
| 29 |
A" |
2961 |
2925 |
6.39 |
|
|
|
| 30 |
A" |
1448 |
1430 |
7.12 |
|
|
|
| 31 |
A" |
1421 |
1403 |
11.07 |
|
|
|
| 32 |
A" |
1277 |
1261 |
0.09 |
|
|
|
| 33 |
A" |
1204 |
1189 |
0.02 |
|
|
|
| 34 |
A" |
1096 |
1083 |
0.71 |
|
|
|
| 35 |
A" |
937 |
926 |
0.82 |
|
|
|
| 36 |
A" |
798 |
788 |
0.80 |
|
|
|
| 37 |
A" |
698 |
689 |
3.99 |
|
|
|
| 38 |
A" |
458 |
452 |
0.01 |
|
|
|
| 39 |
A" |
238 |
235 |
0.05 |
|
|
|
| 40 |
A" |
86 |
84 |
0.07 |
|
|
|
| 41 |
A" |
67 |
66 |
0.79 |
|
|
|
| 42 |
A" |
49i |
49i |
0.41 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 29862.3 cm
-1
Scaled (by 0.9879) Zero Point Vibrational Energy (zpe) 29500.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/Def2TZVPP
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-2.421 |
1.356 |
0.000 |
| C2 |
-1.469 |
0.158 |
0.000 |
| C3 |
0.000 |
0.577 |
0.000 |
| C4 |
0.991 |
-0.579 |
0.000 |
| C5 |
2.462 |
-0.211 |
0.000 |
| O6 |
0.626 |
-1.745 |
0.000 |
| H7 |
-3.471 |
1.032 |
0.000 |
| H8 |
-2.269 |
1.989 |
0.887 |
| H9 |
-2.269 |
1.989 |
-0.887 |
| H10 |
-1.658 |
-0.480 |
0.876 |
| H11 |
-1.658 |
-0.480 |
-0.876 |
| H12 |
0.226 |
1.214 |
-0.874 |
| H13 |
0.226 |
1.214 |
0.874 |
| H14 |
3.075 |
-1.119 |
0.000 |
| H15 |
2.704 |
0.399 |
-0.883 |
| H16 |
2.704 |
0.399 |
0.883 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
| C1 | | 1.5307 | 2.5435 | 3.9227 | 5.1290 | 4.3472 | 1.0985 | 1.1001 | 1.1001 | 2.1724 | 2.1724 | 2.7916 | 2.7916 | 6.0277 | 5.2880 | 5.2880 |
C2 | 1.5307 | | 1.5275 | 2.5680 | 3.9486 | 2.8296 | 2.1845 | 2.1860 | 2.1860 | 1.0995 | 1.0995 | 2.1799 | 2.1799 | 4.7196 | 4.2719 | 4.2719 | C3 | 2.5435 | 1.5275 | | 1.5223 | 2.5855 | 2.4042 | 3.5005 | 2.8155 | 2.8155 | 2.1523 | 2.1523 | 1.1048 | 1.1048 | 3.5115 | 2.8500 | 2.8500 | C4 | 3.9227 | 2.5680 | 1.5223 | | 1.5163 | 1.2220 | 4.7439 | 4.2431 | 4.2431 | 2.7922 | 2.7922 | 2.1355 | 2.1355 | 2.1525 | 2.1604 | 2.1604 | C5 | 5.1290 | 3.9486 | 2.5855 | 1.5163 | | 2.3925 | 6.0622 | 5.2925 | 5.2925 | 4.2212 | 4.2212 | 2.7919 | 2.7919 | 1.0951 | 1.1001 | 1.1001 | O6 | 4.3472 | 2.8296 | 2.4042 | 1.2220 | 2.3925 | | 4.9489 | 4.8064 | 4.8064 | 2.7537 | 2.7537 | 3.1103 | 3.1103 | 2.5274 | 3.1131 | 3.1131 | H7 | 1.0985 | 2.1845 | 3.5005 | 4.7439 | 6.0622 | 4.9489 | | 1.7741 | 1.7741 | 2.5174 | 2.5174 | 3.8033 | 3.8033 | 6.8900 | 6.2696 | 6.2696 | H8 | 1.1001 | 2.1860 | 2.8155 | 4.2431 | 5.2925 | 4.8064 | 1.7741 | | 1.7743 | 2.5428 | 3.0939 | 3.1507 | 2.6125 | 6.2448 | 5.5125 | 5.2205 | H9 | 1.1001 | 2.1860 | 2.8155 | 4.2431 | 5.2925 | 4.8064 | 1.7741 | 1.7743 | | 3.0939 | 2.5428 | 2.6125 | 3.1507 | 6.2448 | 5.2205 | 5.5125 | H10 | 2.1724 | 1.0995 | 2.1523 | 2.7922 | 4.2212 | 2.7537 | 2.5174 | 2.5428 | 3.0939 | | 1.7510 | 3.0789 | 2.5336 | 4.8555 | 4.7846 | 4.4497 | H11 | 2.1724 | 1.0995 | 2.1523 | 2.7922 | 4.2212 | 2.7537 | 2.5174 | 3.0939 | 2.5428 | 1.7510 | | 2.5336 | 3.0789 | 4.8555 | 4.4497 | 4.7846 | H12 | 2.7916 | 2.1799 | 1.1048 | 2.1355 | 2.7919 | 3.1103 | 3.8033 | 3.1507 | 2.6125 | 3.0789 | 2.5336 | | 1.7480 | 3.7842 | 2.6082 | 3.1449 | H13 | 2.7916 | 2.1799 | 1.1048 | 2.1355 | 2.7919 | 3.1103 | 3.8033 | 2.6125 | 3.1507 | 2.5336 | 3.0789 | 1.7480 | | 3.7842 | 3.1449 | 2.6082 | H14 | 6.0277 | 4.7196 | 3.5115 | 2.1525 | 1.0951 | 2.5274 | 6.8900 | 6.2448 | 6.2448 | 4.8555 | 4.8555 | 3.7842 | 3.7842 | | 1.7949 | 1.7949 | H15 | 5.2880 | 4.2719 | 2.8500 | 2.1604 | 1.1001 | 3.1131 | 6.2696 | 5.5125 | 5.2205 | 4.7846 | 4.4497 | 2.6082 | 3.1449 | 1.7949 | | 1.7663 | H16 | 5.2880 | 4.2719 | 2.8500 | 2.1604 | 1.1001 | 3.1131 | 6.2696 | 5.2205 | 5.5125 | 4.4497 | 4.7846 | 3.1449 | 2.6082 | 1.7949 | 1.7663 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
112.551 |
|
C1 |
C2 |
H10 |
110.293 |
| C1 |
C2 |
H11 |
110.293 |
|
C2 |
C1 |
H7 |
111.313 |
| C2 |
C1 |
H8 |
111.335 |
|
C2 |
C1 |
H9 |
111.335 |
| C2 |
C3 |
C4 |
114.711 |
|
C2 |
C3 |
H12 |
110.798 |
| C2 |
C3 |
H13 |
110.798 |
|
C3 |
C2 |
H10 |
108.948 |
| C3 |
C2 |
H11 |
108.948 |
|
C3 |
C4 |
C5 |
116.623 |
| C3 |
C4 |
O6 |
121.965 |
|
C4 |
C3 |
H12 |
107.701 |
| C4 |
C3 |
H13 |
107.701 |
|
C4 |
C5 |
H14 |
109.980 |
| C4 |
C5 |
H15 |
110.311 |
|
C4 |
C5 |
H16 |
110.311 |
| C5 |
C4 |
O6 |
121.412 |
|
H7 |
C1 |
H8 |
107.583 |
| H7 |
C1 |
H9 |
107.583 |
|
H8 |
C1 |
H9 |
107.492 |
| H10 |
C2 |
H11 |
105.556 |
|
H12 |
C3 |
H13 |
104.575 |
| H14 |
C5 |
H15 |
109.697 |
|
H14 |
C5 |
H16 |
109.697 |
| H15 |
C5 |
H16 |
106.791 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.252 |
|
|
|
| 2 |
C |
-0.065 |
|
|
|
| 3 |
C |
-0.110 |
|
|
|
| 4 |
C |
0.150 |
|
|
|
| 5 |
C |
-0.232 |
|
|
|
| 6 |
O |
-0.258 |
|
|
|
| 7 |
H |
0.084 |
|
|
|
| 8 |
H |
0.072 |
|
|
|
| 9 |
H |
0.072 |
|
|
|
| 10 |
H |
0.065 |
|
|
|
| 11 |
H |
0.065 |
|
|
|
| 12 |
H |
0.066 |
|
|
|
| 13 |
H |
0.066 |
|
|
|
| 14 |
H |
0.100 |
|
|
|
| 15 |
H |
0.087 |
|
|
|
| 16 |
H |
0.087 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.674 |
2.590 |
0.000 |
2.676 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-35.891 |
1.461 |
0.000 |
| y |
1.461 |
-44.510 |
0.000 |
| z |
0.000 |
0.000 |
-37.517 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.123 |
1.461 |
0.000 |
| y |
1.461 |
-7.806 |
0.000 |
| z |
0.000 |
0.000 |
2.683 |
|
| Polar |
|---|
| 3z2-r2 | 5.366 |
|---|
| x2-y2 | 8.619 |
|---|
| xy | 1.461 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.743 |
-0.550 |
0.000 |
| y |
-0.550 |
10.146 |
0.000 |
| z |
0.000 |
0.000 |
8.006 |
<r2> (average value of r
2) Å
2
| <r2> |
229.806 |
| (<r2>)1/2 |
15.159 |