Vibrational Frequencies calculated at PBEPBE/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3391 |
3350 |
1.65 |
|
|
|
| 2 |
A |
3053 |
3016 |
19.72 |
|
|
|
| 3 |
A |
3029 |
2992 |
36.76 |
|
|
|
| 4 |
A |
3000 |
2964 |
48.55 |
|
|
|
| 5 |
A |
2995 |
2959 |
45.47 |
|
|
|
| 6 |
A |
2988 |
2952 |
61.55 |
|
|
|
| 7 |
A |
2983 |
2947 |
50.40 |
|
|
|
| 8 |
A |
2960 |
2925 |
33.58 |
|
|
|
| 9 |
A |
2944 |
2908 |
42.87 |
|
|
|
| 10 |
A |
2940 |
2904 |
14.67 |
|
|
|
| 11 |
A |
2935 |
2899 |
27.20 |
|
|
|
| 12 |
A |
2931 |
2896 |
31.94 |
|
|
|
| 13 |
A |
2908 |
2873 |
12.79 |
|
|
|
| 14 |
A |
1451 |
1434 |
5.60 |
|
|
|
| 15 |
A |
1445 |
1427 |
7.21 |
|
|
|
| 16 |
A |
1443 |
1426 |
5.23 |
|
|
|
| 17 |
A |
1434 |
1416 |
12.60 |
|
|
|
| 18 |
A |
1428 |
1411 |
5.41 |
|
|
|
| 19 |
A |
1424 |
1407 |
1.60 |
|
|
|
| 20 |
A |
1415 |
1398 |
9.34 |
|
|
|
| 21 |
A |
1356 |
1339 |
10.60 |
|
|
|
| 22 |
A |
1340 |
1324 |
1.54 |
|
|
|
| 23 |
A |
1330 |
1314 |
4.55 |
|
|
|
| 24 |
A |
1326 |
1310 |
1.29 |
|
|
|
| 25 |
A |
1316 |
1300 |
0.20 |
|
|
|
| 26 |
A |
1312 |
1296 |
2.14 |
|
|
|
| 27 |
A |
1287 |
1272 |
0.97 |
|
|
|
| 28 |
A |
1257 |
1242 |
3.35 |
|
|
|
| 29 |
A |
1240 |
1225 |
4.07 |
|
|
|
| 30 |
A |
1202 |
1188 |
8.06 |
|
|
|
| 31 |
A |
1155 |
1141 |
5.15 |
|
|
|
| 32 |
A |
1137 |
1123 |
10.55 |
|
|
|
| 33 |
A |
1099 |
1086 |
19.42 |
|
|
|
| 34 |
A |
1080 |
1066 |
3.83 |
|
|
|
| 35 |
A |
1067 |
1054 |
5.07 |
|
|
|
| 36 |
A |
1041 |
1029 |
4.72 |
|
|
|
| 37 |
A |
962 |
951 |
4.70 |
|
|
|
| 38 |
A |
950 |
938 |
0.25 |
|
|
|
| 39 |
A |
936 |
925 |
1.37 |
|
|
|
| 40 |
A |
894 |
883 |
10.39 |
|
|
|
| 41 |
A |
858 |
848 |
2.08 |
|
|
|
| 42 |
A |
835 |
825 |
0.23 |
|
|
|
| 43 |
A |
811 |
801 |
25.77 |
|
|
|
| 44 |
A |
782 |
773 |
8.37 |
|
|
|
| 45 |
A |
713 |
704 |
73.63 |
|
|
|
| 46 |
A |
542 |
536 |
1.62 |
|
|
|
| 47 |
A |
456 |
451 |
3.95 |
|
|
|
| 48 |
A |
445 |
439 |
2.14 |
|
|
|
| 49 |
A |
416 |
411 |
1.11 |
|
|
|
| 50 |
A |
320 |
316 |
0.71 |
|
|
|
| 51 |
A |
307 |
303 |
0.88 |
|
|
|
| 52 |
A |
238 |
235 |
2.18 |
|
|
|
| 53 |
A |
208 |
206 |
0.76 |
|
|
|
| 54 |
A |
147 |
146 |
0.98 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 39730.5 cm
-1
Scaled (by 0.9879) Zero Point Vibrational Energy (zpe) 39249.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
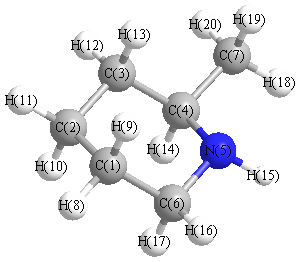
Charges from optimized geometry at PBEPBE/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.097 |
|
|
|
| 2 |
C |
-0.097 |
|
|
|
| 3 |
C |
-0.111 |
|
|
|
| 4 |
C |
0.090 |
|
|
|
| 5 |
N |
-0.207 |
|
|
|
| 6 |
C |
-0.078 |
|
|
|
| 7 |
C |
-0.274 |
|
|
|
| 8 |
H |
0.048 |
|
|
|
| 9 |
H |
0.043 |
|
|
|
| 10 |
H |
0.052 |
|
|
|
| 11 |
H |
0.061 |
|
|
|
| 12 |
H |
0.048 |
|
|
|
| 13 |
H |
0.036 |
|
|
|
| 14 |
H |
0.038 |
|
|
|
| 15 |
H |
0.101 |
|
|
|
| 16 |
H |
0.069 |
|
|
|
| 17 |
H |
0.052 |
|
|
|
| 18 |
H |
0.092 |
|
|
|
| 19 |
H |
0.062 |
|
|
|
| 20 |
H |
0.073 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.446 |
0.752 |
0.484 |
0.999 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.432 |
1.591 |
0.285 |
| y |
1.591 |
-48.654 |
-1.464 |
| z |
0.285 |
-1.464 |
-44.913 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
0.351 |
1.591 |
0.285 |
| y |
1.591 |
-2.981 |
-1.464 |
| z |
0.285 |
-1.464 |
2.630 |
|
| Polar |
|---|
| 3z2-r2 | 5.260 |
|---|
| x2-y2 | 2.222 |
|---|
| xy | 1.591 |
|---|
| xz | 0.285 |
|---|
| yz | -1.464 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
13.825 |
0.176 |
0.076 |
| y |
0.176 |
12.116 |
-0.079 |
| z |
0.076 |
-0.079 |
10.842 |
<r2> (average value of r
2) Å
2
| <r2> |
232.610 |
| (<r2>)1/2 |
15.252 |