Jump to
S1C2
Energy calculated at PBEPBE/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -189.096876 |
| Energy at 298.15K | -189.104177 |
| HF Energy | -189.096876 |
| Nuclear repulsion energy | 121.896995 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3081 |
3043 |
0.41 |
|
|
|
| 2 |
A1 |
2942 |
2907 |
12.92 |
|
|
|
| 3 |
A1 |
1577 |
1558 |
14.34 |
|
|
|
| 4 |
A1 |
1417 |
1400 |
0.23 |
|
|
|
| 5 |
A1 |
1333 |
1317 |
25.08 |
|
|
|
| 6 |
A1 |
1048 |
1036 |
5.68 |
|
|
|
| 7 |
A1 |
838 |
827 |
0.32 |
|
|
|
| 8 |
A1 |
354 |
350 |
1.16 |
|
|
|
| 9 |
A2 |
3014 |
2978 |
0.00 |
|
|
|
| 10 |
A2 |
1423 |
1406 |
0.00 |
|
|
|
| 11 |
A2 |
1040 |
1028 |
0.00 |
|
|
|
| 12 |
A2 |
470 |
464 |
0.00 |
|
|
|
| 13 |
A2 |
99 |
98 |
0.00 |
|
|
|
| 14 |
B1 |
3004 |
2968 |
26.36 |
|
|
|
| 15 |
B1 |
1446 |
1429 |
24.03 |
|
|
|
| 16 |
B1 |
884 |
874 |
2.16 |
|
|
|
| 17 |
B1 |
177 |
175 |
0.03 |
|
|
|
| 18 |
B2 |
3080 |
3043 |
28.16 |
|
|
|
| 19 |
B2 |
2945 |
2909 |
1.55 |
|
|
|
| 20 |
B2 |
1406 |
1389 |
13.32 |
|
|
|
| 21 |
B2 |
1321 |
1305 |
0.70 |
|
|
|
| 22 |
B2 |
1130 |
1116 |
12.71 |
|
|
|
| 23 |
B2 |
900 |
889 |
24.28 |
|
|
|
| 24 |
B2 |
611 |
604 |
0.06 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 17770.1 cm
-1
Scaled (by 0.9879) Zero Point Vibrational Energy (zpe) 17555.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/Def2TZVPP
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.622 |
-0.786 |
| N2 |
0.000 |
-0.622 |
-0.786 |
| C3 |
0.000 |
1.355 |
0.502 |
| C4 |
0.000 |
-1.355 |
0.502 |
| H5 |
0.000 |
2.424 |
0.275 |
| H6 |
0.000 |
-2.424 |
0.275 |
| H7 |
-0.887 |
1.101 |
1.107 |
| H8 |
0.887 |
1.101 |
1.107 |
| H9 |
0.887 |
-1.101 |
1.107 |
| H10 |
-0.887 |
-1.101 |
1.107 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
C3 |
C4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| N1 | | 1.2437 | 1.4820 | 2.3592 | 2.0909 | 3.2249 | 2.1444 | 2.1444 | 2.7088 | 2.7088 |
N2 | 1.2437 | | 2.3592 | 1.4820 | 3.2249 | 2.0909 | 2.7088 | 2.7088 | 2.1444 | 2.1444 | C3 | 1.4820 | 2.3592 | | 2.7091 | 1.0928 | 3.7849 | 1.1028 | 1.1028 | 2.6800 | 2.6800 | C4 | 2.3592 | 1.4820 | 2.7091 | | 3.7849 | 1.0928 | 2.6800 | 2.6800 | 1.1028 | 1.1028 | H5 | 2.0909 | 3.2249 | 1.0928 | 3.7849 | | 4.8471 | 1.7964 | 1.7964 | 3.7284 | 3.7284 | H6 | 3.2249 | 2.0909 | 3.7849 | 1.0928 | 4.8471 | | 3.7284 | 3.7284 | 1.7964 | 1.7964 | H7 | 2.1444 | 2.7088 | 1.1028 | 2.6800 | 1.7964 | 3.7284 | | 1.7735 | 2.8276 | 2.2022 | H8 | 2.1444 | 2.7088 | 1.1028 | 2.6800 | 1.7964 | 3.7284 | 1.7735 | | 2.2022 | 2.8276 | H9 | 2.7088 | 2.1444 | 2.6800 | 1.1028 | 3.7284 | 1.7964 | 2.8276 | 2.2022 | | 1.7735 | H10 | 2.7088 | 2.1444 | 2.6800 | 1.1028 | 3.7284 | 1.7964 | 2.2022 | 2.8276 | 1.7735 | |
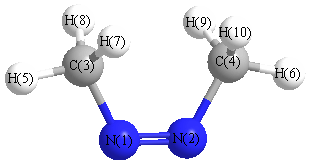 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
N2 |
C4 |
119.631 |
|
N1 |
C3 |
H5 |
107.634 |
| N1 |
C3 |
H7 |
111.278 |
|
N1 |
C3 |
H8 |
111.278 |
| N2 |
N1 |
C3 |
119.631 |
|
N2 |
C4 |
H6 |
107.634 |
| N2 |
C4 |
H9 |
111.278 |
|
N2 |
C4 |
H10 |
111.278 |
| H5 |
C3 |
H7 |
109.802 |
|
H5 |
C3 |
H8 |
109.802 |
| H6 |
C4 |
H9 |
109.802 |
|
H6 |
C4 |
H10 |
109.802 |
| H7 |
C3 |
H8 |
107.051 |
|
H9 |
C4 |
H10 |
107.051 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.100 |
|
|
|
| 2 |
N |
-0.100 |
|
|
|
| 3 |
C |
-0.211 |
|
|
|
| 4 |
C |
-0.211 |
|
|
|
| 5 |
H |
0.114 |
|
|
|
| 6 |
H |
0.114 |
|
|
|
| 7 |
H |
0.099 |
|
|
|
| 8 |
H |
0.099 |
|
|
|
| 9 |
H |
0.099 |
|
|
|
| 10 |
H |
0.099 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
3.122 |
3.122 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-24.802 |
0.000 |
0.000 |
| y |
0.000 |
-23.867 |
0.000 |
| z |
0.000 |
0.000 |
-29.249 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.756 |
0.000 |
0.000 |
| y |
0.000 |
3.158 |
0.000 |
| z |
0.000 |
0.000 |
-4.914 |
|
| Polar |
|---|
| 3z2-r2 | -9.829 |
|---|
| x2-y2 | -0.935 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.946 |
0.000 |
0.000 |
| y |
0.000 |
8.289 |
0.000 |
| z |
0.000 |
0.000 |
6.109 |
<r2> (average value of r
2) Å
2
| <r2> |
80.342 |
| (<r2>)1/2 |
8.963 |
Jump to
S1C1
Energy calculated at PBEPBE/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -189.096876 |
| Energy at 298.15K | -189.104176 |
| HF Energy | -189.096876 |
| Nuclear repulsion energy | 121.899391 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3081 |
3043 |
0.42 |
|
|
|
| 2 |
A |
3014 |
2978 |
0.00 |
|
|
|
| 3 |
A |
2942 |
2907 |
12.92 |
|
|
|
| 4 |
A |
1577 |
1558 |
14.33 |
|
|
|
| 5 |
A |
1423 |
1406 |
0.00 |
|
|
|
| 6 |
A |
1417 |
1400 |
0.23 |
|
|
|
| 7 |
A |
1333 |
1317 |
25.08 |
|
|
|
| 8 |
A |
1048 |
1036 |
5.69 |
|
|
|
| 9 |
A |
1040 |
1028 |
0.00 |
|
|
|
| 10 |
A |
838 |
828 |
0.32 |
|
|
|
| 11 |
A |
470 |
464 |
0.00 |
|
|
|
| 12 |
A |
354 |
350 |
1.16 |
|
|
|
| 13 |
A |
99 |
97 |
0.00 |
|
|
|
| 14 |
B |
3080 |
3043 |
28.17 |
|
|
|
| 15 |
B |
3004 |
2968 |
26.37 |
|
|
|
| 16 |
B |
2945 |
2909 |
1.56 |
|
|
|
| 17 |
B |
1446 |
1429 |
24.03 |
|
|
|
| 18 |
B |
1406 |
1389 |
13.31 |
|
|
|
| 19 |
B |
1321 |
1305 |
0.71 |
|
|
|
| 20 |
B |
1130 |
1116 |
12.70 |
|
|
|
| 21 |
B |
900 |
890 |
24.26 |
|
|
|
| 22 |
B |
884 |
874 |
2.16 |
|
|
|
| 23 |
B |
611 |
604 |
0.06 |
|
|
|
| 24 |
B |
176 |
174 |
0.03 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 17770.1 cm
-1
Scaled (by 0.9879) Zero Point Vibrational Energy (zpe) 17555.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/Def2TZVPP
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
-0.015 |
0.622 |
-0.777 |
| N2 |
0.015 |
-0.622 |
-0.777 |
| C3 |
0.015 |
1.373 |
0.496 |
| C4 |
-0.015 |
-1.373 |
0.496 |
| H5 |
0.052 |
2.441 |
0.257 |
| H6 |
-0.052 |
-2.441 |
0.257 |
| H7 |
-0.895 |
1.172 |
1.084 |
| H8 |
0.884 |
1.108 |
1.120 |
| H9 |
0.895 |
-1.172 |
1.084 |
| H10 |
-0.884 |
-1.108 |
1.120 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
C3 |
C4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| N1 | | 1.2436 | 1.4790 | 2.3666 | 2.0935 | 3.2324 | 2.1309 | 2.1550 | 2.7401 | 2.7101 |
N2 | 1.2436 | | 2.3666 | 1.4790 | 3.2324 | 2.0935 | 2.7401 | 2.7101 | 2.1309 | 2.1550 | C3 | 1.4790 | 2.3666 | | 2.7466 | 1.0946 | 3.8220 | 1.1020 | 1.1020 | 2.7561 | 2.7116 | C4 | 2.3666 | 1.4790 | 2.7466 | | 3.8220 | 1.0946 | 2.7561 | 2.7116 | 1.1020 | 1.1020 | H5 | 2.0935 | 3.2324 | 1.0946 | 3.8220 | | 4.8825 | 1.7863 | 1.7927 | 3.8006 | 3.7700 | H6 | 3.2324 | 2.0935 | 3.8220 | 1.0946 | 4.8825 | | 3.8006 | 3.7700 | 1.7863 | 1.7927 | H7 | 2.1309 | 2.7401 | 1.1020 | 2.7561 | 1.7863 | 3.8006 | | 1.7805 | 2.9488 | 2.2798 | H8 | 2.1550 | 2.7101 | 1.1020 | 2.7116 | 1.7927 | 3.7700 | 1.7805 | | 2.2798 | 2.8347 | H9 | 2.7401 | 2.1309 | 2.7561 | 1.1020 | 3.8006 | 1.7863 | 2.9488 | 2.2798 | | 1.7805 | H10 | 2.7101 | 2.1550 | 2.7116 | 1.1020 | 3.7700 | 1.7927 | 2.2798 | 2.8347 | 1.7805 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
N2 |
C4 |
120.500 |
|
N1 |
C3 |
H5 |
107.940 |
| N1 |
C3 |
H7 |
110.453 |
|
N1 |
C3 |
H8 |
112.397 |
| N2 |
N1 |
C3 |
120.500 |
|
N2 |
C4 |
H6 |
107.940 |
| N2 |
C4 |
H9 |
110.453 |
|
N2 |
C4 |
H10 |
112.397 |
| H5 |
C3 |
H7 |
108.824 |
|
H5 |
C3 |
H8 |
109.398 |
| H6 |
C4 |
H9 |
108.824 |
|
H6 |
C4 |
H10 |
109.398 |
| H7 |
C3 |
H8 |
107.776 |
|
H9 |
C4 |
H10 |
107.776 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.100 |
|
|
|
| 2 |
N |
-0.100 |
|
|
|
| 3 |
C |
-0.211 |
|
|
|
| 4 |
C |
-0.211 |
|
|
|
| 5 |
H |
0.114 |
|
|
|
| 6 |
H |
0.114 |
|
|
|
| 7 |
H |
0.099 |
|
|
|
| 8 |
H |
0.099 |
|
|
|
| 9 |
H |
0.099 |
|
|
|
| 10 |
H |
0.099 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
3.122 |
3.122 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-24.802 |
0.000 |
0.000 |
| y |
0.000 |
-23.866 |
0.000 |
| z |
0.000 |
0.000 |
-29.248 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.755 |
0.000 |
0.000 |
| y |
0.000 |
3.159 |
0.000 |
| z |
0.000 |
0.000 |
-4.914 |
|
| Polar |
|---|
| 3z2-r2 | -9.828 |
|---|
| x2-y2 | -0.935 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.946 |
0.000 |
0.000 |
| y |
0.000 |
8.289 |
0.000 |
| z |
0.000 |
0.000 |
6.109 |
<r2> (average value of r
2) Å
2
| <r2> |
80.339 |
| (<r2>)1/2 |
8.963 |