Jump to
S1C2
Energy calculated at PBEPBE/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -941.085664 |
| Energy at 298.15K | -941.087466 |
| HF Energy | -941.085664 |
| Nuclear repulsion energy | 233.708130 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
1721 |
1701 |
581.00 |
|
|
|
| 2 |
A1 |
917 |
906 |
77.66 |
|
|
|
| 3 |
A1 |
736 |
727 |
64.72 |
|
|
|
| 4 |
A1 |
439 |
434 |
65.98 |
|
|
|
| 5 |
B1 |
789 |
779 |
11.88 |
|
|
|
| 6 |
B1 |
111 |
109 |
17.21 |
|
|
|
| 7 |
B2 |
1025 |
1013 |
329.48 |
|
|
|
| 8 |
B2 |
632 |
625 |
0.94 |
|
|
|
| 9 |
B2 |
359 |
355 |
11.67 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3365.2 cm
-1
Scaled (by 0.9879) Zero Point Vibrational Energy (zpe) 3324.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/Def2TZVPP
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
-1.062 |
| O2 |
0.000 |
0.000 |
-2.282 |
| Ca3 |
0.000 |
0.000 |
1.489 |
| O4 |
0.000 |
1.125 |
-0.322 |
| O5 |
0.000 |
-1.125 |
-0.322 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
Ca3 |
O4 |
O5 |
| C1 | | 1.2195 | 2.5512 | 1.3465 | 1.3465 |
O2 | 1.2195 | | 3.7707 | 2.2598 | 2.2598 | Ca3 | 2.5512 | 3.7707 | | 2.1314 | 2.1314 | O4 | 1.3465 | 2.2598 | 2.1314 | | 2.2492 | O5 | 1.3465 | 2.2598 | 2.1314 | 2.2492 | |
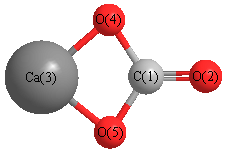 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O2 |
C1 |
Ca3 |
180.000 |
|
O2 |
C1 |
O4 |
123.368 |
| O2 |
C1 |
O5 |
123.368 |
|
Ca3 |
C1 |
O4 |
56.632 |
| Ca3 |
C1 |
O5 |
56.632 |
|
O4 |
C1 |
O5 |
113.265 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.318 |
|
|
|
| 2 |
O |
-0.315 |
|
|
|
| 3 |
Ca |
1.113 |
|
|
|
| 4 |
O |
-0.558 |
|
|
|
| 5 |
O |
-0.558 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
12.562 |
12.562 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.116 |
0.000 |
0.000 |
| y |
0.000 |
-41.993 |
0.000 |
| z |
0.000 |
0.000 |
-29.370 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.566 |
0.000 |
0.000 |
| y |
0.000 |
-11.250 |
0.000 |
| z |
0.000 |
0.000 |
7.684 |
|
| Polar |
|---|
| 3z2-r2 | 15.369 |
|---|
| x2-y2 | 9.877 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.863 |
0.000 |
0.000 |
| y |
0.000 |
6.221 |
0.000 |
| z |
0.000 |
0.000 |
10.474 |
<r2> (average value of r
2) Å
2
| <r2> |
131.971 |
| (<r2>)1/2 |
11.488 |
Jump to
S1C1
Energy calculated at PBEPBE/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -941.085663 |
| Energy at 298.15K | -941.087466 |
| HF Energy | -941.085663 |
| Nuclear repulsion energy | 233.686753 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
1720 |
1699 |
580.93 |
|
|
|
| 2 |
A' |
1025 |
1012 |
329.35 |
|
|
|
| 3 |
A' |
917 |
905 |
77.87 |
|
|
|
| 4 |
A' |
736 |
727 |
64.63 |
|
|
|
| 5 |
A' |
632 |
624 |
0.99 |
|
|
|
| 6 |
A' |
439 |
434 |
65.99 |
|
|
|
| 7 |
A' |
360 |
355 |
11.65 |
|
|
|
| 8 |
A" |
789 |
780 |
11.87 |
|
|
|
| 9 |
A" |
111 |
110 |
17.22 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3364.2 cm
-1
Scaled (by 0.9879) Zero Point Vibrational Energy (zpe) 3323.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/Def2TZVPP
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
1.065 |
0.000 |
| O2 |
0.020 |
2.285 |
0.000 |
| Ca3 |
-0.009 |
-1.464 |
0.000 |
| O4 |
-1.129 |
0.300 |
0.000 |
| O5 |
1.130 |
0.277 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
Ca3 |
O4 |
O5 |
| C1 | | 1.2201 | 2.5287 | 1.3632 | 1.3776 |
O2 | 1.2201 | | 3.7487 | 2.2930 | 2.2941 | Ca3 | 2.5287 | 3.7487 | | 2.0897 | 2.0800 | O4 | 1.3632 | 2.2930 | 2.0897 | | 2.2588 | O5 | 1.3776 | 2.2941 | 2.0800 | 2.2588 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O2 |
C1 |
Ca3 |
179.241 |
|
O2 |
C1 |
O4 |
125.063 |
| O2 |
C1 |
O5 |
123.936 |
|
Ca3 |
C1 |
O4 |
55.696 |
| Ca3 |
C1 |
O5 |
55.305 |
|
O4 |
C1 |
O5 |
111.001 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.318 |
|
|
|
| 2 |
O |
-0.315 |
|
|
|
| 3 |
Ca |
1.113 |
|
|
|
| 4 |
O |
-0.558 |
|
|
|
| 5 |
O |
-0.559 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.011 |
-12.562 |
0.000 |
12.562 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-41.995 |
0.004 |
0.000 |
| y |
0.004 |
-29.369 |
0.000 |
| z |
0.000 |
0.000 |
-32.117 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-11.252 |
0.004 |
0.000 |
| y |
0.004 |
7.687 |
0.000 |
| z |
0.000 |
0.000 |
3.565 |
|
| Polar |
|---|
| 3z2-r2 | 7.130 |
|---|
| x2-y2 | -12.626 |
|---|
| xy | 0.004 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.222 |
0.004 |
0.000 |
| y |
0.004 |
10.475 |
0.000 |
| z |
0.000 |
0.000 |
3.864 |
<r2> (average value of r
2) Å
2
| <r2> |
131.989 |
| (<r2>)1/2 |
11.489 |