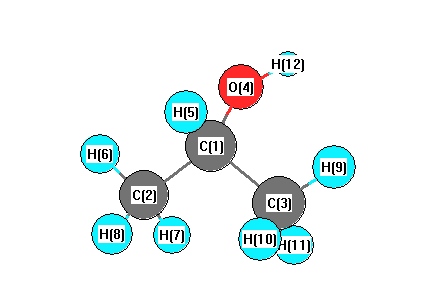
Vibrational Frequencies calculated at PBEPBE/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3714 |
3669 |
10.98 |
|
|
|
| 2 |
A |
3058 |
3021 |
20.96 |
|
|
|
| 3 |
A |
3050 |
3013 |
39.17 |
|
|
|
| 4 |
A |
3041 |
3004 |
8.31 |
|
|
|
| 5 |
A |
3028 |
2991 |
30.64 |
|
|
|
| 6 |
A |
2975 |
2939 |
13.15 |
|
|
|
| 7 |
A |
2959 |
2923 |
21.87 |
|
|
|
| 8 |
A |
2888 |
2853 |
54.57 |
|
|
|
| 9 |
A |
1455 |
1438 |
7.54 |
|
|
|
| 10 |
A |
1445 |
1427 |
5.35 |
|
|
|
| 11 |
A |
1434 |
1416 |
2.52 |
|
|
|
| 12 |
A |
1429 |
1412 |
0.66 |
|
|
|
| 13 |
A |
1375 |
1358 |
23.04 |
|
|
|
| 14 |
A |
1357 |
1341 |
19.53 |
|
|
|
| 15 |
A |
1338 |
1322 |
2.76 |
|
|
|
| 16 |
A |
1321 |
1305 |
9.20 |
|
|
|
| 17 |
A |
1238 |
1223 |
41.71 |
|
|
|
| 18 |
A |
1146 |
1132 |
25.76 |
|
|
|
| 19 |
A |
1117 |
1103 |
29.58 |
|
|
|
| 20 |
A |
1058 |
1045 |
21.91 |
|
|
|
| 21 |
A |
932 |
920 |
39.36 |
|
|
|
| 22 |
A |
917 |
906 |
5.97 |
|
|
|
| 23 |
A |
896 |
885 |
0.28 |
|
|
|
| 24 |
A |
804 |
795 |
5.65 |
|
|
|
| 25 |
A |
466 |
460 |
5.42 |
|
|
|
| 26 |
A |
401 |
397 |
10.40 |
|
|
|
| 27 |
A |
350 |
346 |
2.33 |
|
|
|
| 28 |
A |
282 |
278 |
88.57 |
|
|
|
| 29 |
A |
253 |
250 |
7.07 |
|
|
|
| 30 |
A |
215 |
212 |
5.79 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 22969.1 cm
-1
Scaled (by 0.9879) Zero Point Vibrational Energy (zpe) 22691.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.126 |
|
|
|
| 2 |
C |
-0.249 |
|
|
|
| 3 |
C |
-0.230 |
|
|
|
| 4 |
O |
-0.319 |
|
|
|
| 5 |
H |
0.044 |
|
|
|
| 6 |
H |
0.065 |
|
|
|
| 7 |
H |
0.084 |
|
|
|
| 8 |
H |
0.067 |
|
|
|
| 9 |
H |
0.088 |
|
|
|
| 10 |
H |
0.070 |
|
|
|
| 11 |
H |
0.075 |
|
|
|
| 12 |
H |
0.178 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.095 |
-0.722 |
0.762 |
1.517 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-25.662 |
-2.578 |
-0.249 |
| y |
-2.578 |
-27.331 |
0.937 |
| z |
-0.249 |
0.937 |
-27.160 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.584 |
-2.578 |
-0.249 |
| y |
-2.578 |
-0.920 |
0.937 |
| z |
-0.249 |
0.937 |
-0.664 |
|
| Polar |
|---|
| 3z2-r2 | -1.327 |
|---|
| x2-y2 | 1.670 |
|---|
| xy | -2.578 |
|---|
| xz | -0.249 |
|---|
| yz | 0.937 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.296 |
-0.099 |
-0.033 |
| y |
-0.099 |
6.858 |
-0.006 |
| z |
-0.033 |
-0.006 |
6.190 |
<r2> (average value of r
2) Å
2
| <r2> |
89.885 |
| (<r2>)1/2 |
9.481 |