Jump to
S1C2
Energy calculated at PBEPBE/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -135.041613 |
| Energy at 298.15K | -135.049663 |
| HF Energy | -135.041613 |
| Nuclear repulsion energy | 82.426002 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3395 |
3354 |
2.55 |
|
|
|
| 2 |
A' |
3024 |
2988 |
43.13 |
|
|
|
| 3 |
A' |
2961 |
2926 |
35.90 |
|
|
|
| 4 |
A' |
2953 |
2917 |
22.89 |
|
|
|
| 5 |
A' |
1608 |
1589 |
18.16 |
|
|
|
| 6 |
A' |
1451 |
1434 |
2.86 |
|
|
|
| 7 |
A' |
1433 |
1416 |
0.10 |
|
|
|
| 8 |
A' |
1353 |
1337 |
7.75 |
|
|
|
| 9 |
A' |
1330 |
1314 |
11.63 |
|
|
|
| 10 |
A' |
1118 |
1104 |
7.79 |
|
|
|
| 11 |
A' |
1038 |
1026 |
23.34 |
|
|
|
| 12 |
A' |
870 |
860 |
33.80 |
|
|
|
| 13 |
A' |
826 |
816 |
136.42 |
|
|
|
| 14 |
A' |
389 |
384 |
6.06 |
|
|
|
| 15 |
A" |
3473 |
3431 |
0.01 |
|
|
|
| 16 |
A" |
3031 |
2994 |
49.24 |
|
|
|
| 17 |
A" |
2991 |
2955 |
7.91 |
|
|
|
| 18 |
A" |
1441 |
1424 |
8.13 |
|
|
|
| 19 |
A" |
1347 |
1330 |
0.02 |
|
|
|
| 20 |
A" |
1229 |
1214 |
0.05 |
|
|
|
| 21 |
A" |
976 |
965 |
0.84 |
|
|
|
| 22 |
A" |
755 |
746 |
1.55 |
|
|
|
| 23 |
A" |
277 |
273 |
29.67 |
|
|
|
| 24 |
A" |
246 |
243 |
13.60 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 19757.1 cm
-1
Scaled (by 0.9879) Zero Point Vibrational Energy (zpe) 19518.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/Def2TZVPP
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
-1.313 |
-0.093 |
0.000 |
| C2 |
0.000 |
0.571 |
0.000 |
| C3 |
1.219 |
-0.349 |
0.000 |
| H4 |
2.163 |
0.221 |
0.000 |
| H5 |
1.226 |
-0.999 |
0.888 |
| H6 |
1.226 |
-0.999 |
-0.888 |
| H7 |
0.033 |
1.235 |
-0.878 |
| H8 |
0.033 |
1.235 |
0.878 |
| H9 |
-1.404 |
-0.690 |
0.821 |
| H10 |
-1.404 |
-0.690 |
-0.821 |
Atom - Atom Distances (Å)
| |
N1 |
C2 |
C3 |
H4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| N1 | | 1.4713 | 2.5450 | 3.4898 | 2.8379 | 2.8379 | 2.0849 | 2.0849 | 1.0193 | 1.0193 |
C2 | 1.4713 | | 1.5276 | 2.1910 | 2.1812 | 2.1812 | 1.1015 | 1.1015 | 2.0582 | 2.0582 | C3 | 2.5450 | 1.5276 | | 1.1023 | 1.1005 | 1.1005 | 2.1652 | 2.1652 | 2.7696 | 2.7696 | H4 | 3.4898 | 2.1910 | 1.1023 | | 1.7764 | 1.7764 | 2.5171 | 2.5171 | 3.7716 | 3.7716 | H5 | 2.8379 | 2.1812 | 1.1005 | 1.7764 | | 1.7755 | 3.0878 | 2.5329 | 2.6484 | 3.1511 | H6 | 2.8379 | 2.1812 | 1.1005 | 1.7764 | 1.7755 | | 2.5329 | 3.0878 | 3.1511 | 2.6484 | H7 | 2.0849 | 1.1015 | 2.1652 | 2.5171 | 3.0878 | 2.5329 | | 1.7567 | 2.9427 | 2.4032 | H8 | 2.0849 | 1.1015 | 2.1652 | 2.5171 | 2.5329 | 3.0878 | 1.7567 | | 2.4032 | 2.9427 | H9 | 1.0193 | 2.0582 | 2.7696 | 3.7716 | 2.6484 | 3.1511 | 2.9427 | 2.4032 | | 1.6419 | H10 | 1.0193 | 2.0582 | 2.7696 | 3.7716 | 3.1511 | 2.6484 | 2.4032 | 2.9427 | 1.6419 | |
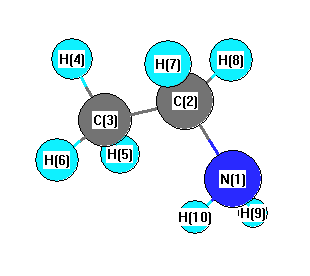 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C2 |
C3 |
116.116 |
|
N1 |
C2 |
H7 |
107.395 |
| N1 |
C2 |
H8 |
107.395 |
|
C2 |
N1 |
H9 |
110.134 |
| C2 |
N1 |
H10 |
110.134 |
|
C2 |
C3 |
H4 |
111.824 |
| C2 |
C3 |
H5 |
111.142 |
|
C2 |
C3 |
H6 |
111.142 |
| C3 |
C2 |
H7 |
109.819 |
|
C3 |
C2 |
H8 |
109.819 |
| H4 |
C3 |
H5 |
107.491 |
|
H4 |
C3 |
H6 |
107.491 |
| H5 |
C3 |
H6 |
107.542 |
|
H7 |
C2 |
H8 |
105.769 |
| H9 |
N1 |
H10 |
107.298 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.326 |
|
|
|
| 2 |
C |
-0.031 |
|
|
|
| 3 |
C |
-0.241 |
|
|
|
| 4 |
H |
0.066 |
|
|
|
| 5 |
H |
0.068 |
|
|
|
| 6 |
H |
0.068 |
|
|
|
| 7 |
H |
0.069 |
|
|
|
| 8 |
H |
0.069 |
|
|
|
| 9 |
H |
0.128 |
|
|
|
| 10 |
H |
0.128 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.862 |
-0.893 |
0.000 |
1.241 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-24.196 |
2.538 |
0.000 |
| y |
2.538 |
-20.739 |
0.000 |
| z |
0.000 |
0.000 |
-19.427 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-4.113 |
2.538 |
0.000 |
| y |
2.538 |
1.073 |
0.000 |
| z |
0.000 |
0.000 |
3.040 |
|
| Polar |
|---|
| 3z2-r2 | 6.080 |
|---|
| x2-y2 | -3.457 |
|---|
| xy | 2.538 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.171 |
0.063 |
0.000 |
| y |
0.063 |
5.423 |
0.000 |
| z |
0.000 |
0.000 |
5.163 |
<r2> (average value of r
2) Å
2
| <r2> |
59.196 |
| (<r2>)1/2 |
7.694 |
Jump to
S1C1
Energy calculated at PBEPBE/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -135.041295 |
| Energy at 298.15K | |
| Nuclear repulsion energy | |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/Def2TZVPP
Geometric Data calculated at PBEPBE/Def2TZVPP
Point Group is Cs
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability