Jump to
S1C2
Energy calculated at PBEPBE/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -232.261117 |
| Energy at 298.15K | |
| HF Energy | -232.261117 |
| Nuclear repulsion energy | 175.435379 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3085 |
3048 |
8.93 |
|
|
|
| 2 |
A' |
3056 |
3019 |
17.34 |
|
|
|
| 3 |
A' |
2984 |
2948 |
21.71 |
|
|
|
| 4 |
A' |
2970 |
2934 |
6.30 |
|
|
|
| 5 |
A' |
2947 |
2911 |
17.65 |
|
|
|
| 6 |
A' |
1727 |
1706 |
139.46 |
|
|
|
| 7 |
A' |
1447 |
1430 |
8.44 |
|
|
|
| 8 |
A' |
1413 |
1396 |
22.23 |
|
|
|
| 9 |
A' |
1393 |
1376 |
5.59 |
|
|
|
| 10 |
A' |
1361 |
1344 |
7.44 |
|
|
|
| 11 |
A' |
1328 |
1312 |
55.35 |
|
|
|
| 12 |
A' |
1312 |
1297 |
14.74 |
|
|
|
| 13 |
A' |
1150 |
1136 |
67.86 |
|
|
|
| 14 |
A' |
1074 |
1061 |
0.83 |
|
|
|
| 15 |
A' |
981 |
969 |
4.52 |
|
|
|
| 16 |
A' |
905 |
894 |
12.92 |
|
|
|
| 17 |
A' |
742 |
733 |
5.27 |
|
|
|
| 18 |
A' |
572 |
565 |
5.89 |
|
|
|
| 19 |
A' |
391 |
386 |
3.65 |
|
|
|
| 20 |
A' |
238 |
235 |
5.20 |
|
|
|
| 21 |
A" |
3058 |
3021 |
18.79 |
|
|
|
| 22 |
A" |
3031 |
2995 |
7.02 |
|
|
|
| 23 |
A" |
2973 |
2937 |
6.44 |
|
|
|
| 24 |
A" |
1440 |
1422 |
8.45 |
|
|
|
| 25 |
A" |
1422 |
1405 |
9.93 |
|
|
|
| 26 |
A" |
1235 |
1220 |
0.01 |
|
|
|
| 27 |
A" |
1087 |
1073 |
0.12 |
|
|
|
| 28 |
A" |
911 |
900 |
3.76 |
|
|
|
| 29 |
A" |
722 |
714 |
2.85 |
|
|
|
| 30 |
A" |
457 |
452 |
0.02 |
|
|
|
| 31 |
A" |
188 |
185 |
0.17 |
|
|
|
| 32 |
A" |
97 |
96 |
0.03 |
|
|
|
| 33 |
A" |
35i |
35i |
0.10 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23829.6 cm
-1
Scaled (by 0.9879) Zero Point Vibrational Energy (zpe) 23541.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/Def2TZVPP
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
1.076 |
1.634 |
0.000 |
| C2 |
0.000 |
0.557 |
0.000 |
| C3 |
0.502 |
-0.885 |
0.000 |
| C4 |
-0.613 |
-1.928 |
0.000 |
| O5 |
-1.183 |
0.830 |
0.000 |
| H6 |
0.609 |
2.624 |
0.000 |
| H7 |
1.723 |
1.531 |
0.883 |
| H8 |
1.723 |
1.531 |
-0.883 |
| H9 |
-0.195 |
-2.943 |
0.000 |
| H10 |
-1.254 |
-1.817 |
0.883 |
| H11 |
-1.254 |
-1.817 |
-0.883 |
| H12 |
1.162 |
-1.010 |
-0.874 |
| H13 |
1.162 |
-1.010 |
0.874 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
O5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.5224 | 2.5841 | 3.9420 | 2.3973 | 1.0947 | 1.0997 | 1.0997 | 4.7498 | 4.2562 | 4.2562 | 2.7862 | 2.7862 |
C2 | 1.5224 | | 1.5270 | 2.5591 | 1.2142 | 2.1555 | 2.1673 | 2.1673 | 3.5048 | 2.8259 | 2.8259 | 2.1377 | 2.1377 | C3 | 2.5841 | 1.5270 | | 1.5265 | 2.4048 | 3.5115 | 2.8476 | 2.8476 | 2.1720 | 2.1749 | 2.1749 | 1.1028 | 1.1028 | C4 | 3.9420 | 2.5591 | 1.5265 | | 2.8165 | 4.7135 | 4.2661 | 4.2661 | 1.0977 | 1.0967 | 1.0967 | 2.1818 | 2.1818 | O5 | 2.3973 | 1.2142 | 2.4048 | 2.8165 | | 2.5356 | 3.1168 | 3.1168 | 3.9003 | 2.7916 | 2.7916 | 3.1067 | 3.1067 | H6 | 1.0947 | 2.1555 | 3.5115 | 4.7135 | 2.5356 | | 1.7935 | 1.7935 | 5.6248 | 4.8964 | 4.8964 | 3.7787 | 3.7787 | H7 | 1.0997 | 2.1673 | 2.8476 | 4.2661 | 3.1168 | 1.7935 | | 1.7658 | 4.9467 | 4.4798 | 4.8154 | 3.1397 | 2.6018 | H8 | 1.0997 | 2.1673 | 2.8476 | 4.2661 | 3.1168 | 1.7935 | 1.7658 | | 4.9467 | 4.8154 | 4.4798 | 2.6018 | 3.1397 | H9 | 4.7498 | 3.5048 | 2.1720 | 1.0977 | 3.9003 | 5.6248 | 4.9467 | 4.9467 | | 1.7802 | 1.7802 | 2.5184 | 2.5184 | H10 | 4.2562 | 2.8259 | 2.1749 | 1.0967 | 2.7916 | 4.8964 | 4.4798 | 4.8154 | 1.7802 | | 1.7665 | 3.0949 | 2.5474 | H11 | 4.2562 | 2.8259 | 2.1749 | 1.0967 | 2.7916 | 4.8964 | 4.8154 | 4.4798 | 1.7802 | 1.7665 | | 2.5474 | 3.0949 | H12 | 2.7862 | 2.1377 | 1.1028 | 2.1818 | 3.1067 | 3.7787 | 3.1397 | 2.6018 | 2.5184 | 3.0949 | 2.5474 | | 1.7488 | H13 | 2.7862 | 2.1377 | 1.1028 | 2.1818 | 3.1067 | 3.7787 | 2.6018 | 3.1397 | 2.5184 | 2.5474 | 3.0949 | 1.7488 | |
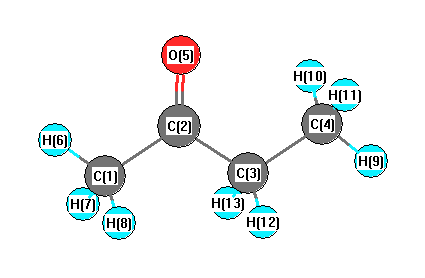 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
115.856 |
|
C1 |
C2 |
O5 |
121.923 |
| C2 |
C1 |
H6 |
109.819 |
|
C2 |
C1 |
H7 |
110.456 |
| C2 |
C1 |
H8 |
110.456 |
|
C2 |
C3 |
C4 |
113.874 |
| C2 |
C3 |
H12 |
107.661 |
|
C2 |
C3 |
H13 |
107.661 |
| C3 |
C2 |
O5 |
122.220 |
|
C3 |
C4 |
H9 |
110.657 |
| C3 |
C4 |
H10 |
110.953 |
|
C3 |
C4 |
H11 |
110.953 |
| C4 |
C3 |
H12 |
111.130 |
|
C4 |
C3 |
H13 |
111.130 |
| H6 |
C1 |
H7 |
109.626 |
|
H6 |
C1 |
H8 |
109.626 |
| H7 |
C1 |
H8 |
106.809 |
|
H9 |
C4 |
H10 |
108.437 |
| H9 |
C4 |
H11 |
108.437 |
|
H10 |
C4 |
H11 |
107.286 |
| H12 |
C3 |
H13 |
104.910 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.229 |
|
|
|
| 2 |
C |
0.148 |
|
|
|
| 3 |
C |
-0.091 |
|
|
|
| 4 |
C |
-0.241 |
|
|
|
| 5 |
O |
-0.257 |
|
|
|
| 6 |
H |
0.101 |
|
|
|
| 7 |
H |
0.087 |
|
|
|
| 8 |
H |
0.087 |
|
|
|
| 9 |
H |
0.080 |
|
|
|
| 10 |
H |
0.087 |
|
|
|
| 11 |
H |
0.087 |
|
|
|
| 12 |
H |
0.071 |
|
|
|
| 13 |
H |
0.071 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.689 |
-0.678 |
0.000 |
2.773 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-34.309 |
2.387 |
0.000 |
| y |
2.387 |
-31.564 |
0.000 |
| z |
0.000 |
0.000 |
-30.914 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.070 |
2.387 |
0.000 |
| y |
2.387 |
1.047 |
0.000 |
| z |
0.000 |
0.000 |
2.022 |
|
| Polar |
|---|
| 3z2-r2 | 4.045 |
|---|
| x2-y2 | -2.745 |
|---|
| xy | 2.387 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.570 |
0.487 |
0.000 |
| y |
0.487 |
9.137 |
0.000 |
| z |
0.000 |
0.000 |
6.478 |
<r2> (average value of r
2) Å
2
| <r2> |
137.998 |
| (<r2>)1/2 |
11.747 |
Jump to
S1C1
Energy calculated at PBEPBE/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -232.261134 |
| Energy at 298.15K | |
| HF Energy | -232.261134 |
| Nuclear repulsion energy | 175.457897 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3086 |
3049 |
8.89 |
|
|
|
| 2 |
A |
3060 |
3023 |
18.67 |
|
|
|
| 3 |
A |
3057 |
3020 |
17.24 |
|
|
|
| 4 |
A |
3032 |
2995 |
7.08 |
|
|
|
| 5 |
A |
2986 |
2949 |
21.84 |
|
|
|
| 6 |
A |
2973 |
2937 |
6.48 |
|
|
|
| 7 |
A |
2971 |
2935 |
6.36 |
|
|
|
| 8 |
A |
2947 |
2912 |
17.48 |
|
|
|
| 9 |
A |
1726 |
1705 |
139.23 |
|
|
|
| 10 |
A |
1448 |
1431 |
8.50 |
|
|
|
| 11 |
A |
1440 |
1423 |
8.51 |
|
|
|
| 12 |
A |
1422 |
1405 |
9.92 |
|
|
|
| 13 |
A |
1413 |
1396 |
22.94 |
|
|
|
| 14 |
A |
1394 |
1377 |
4.80 |
|
|
|
| 15 |
A |
1363 |
1347 |
7.46 |
|
|
|
| 16 |
A |
1330 |
1314 |
55.51 |
|
|
|
| 17 |
A |
1315 |
1299 |
14.84 |
|
|
|
| 18 |
A |
1238 |
1223 |
0.01 |
|
|
|
| 19 |
A |
1150 |
1137 |
67.77 |
|
|
|
| 20 |
A |
1087 |
1074 |
0.11 |
|
|
|
| 21 |
A |
1075 |
1062 |
0.81 |
|
|
|
| 22 |
A |
981 |
969 |
4.48 |
|
|
|
| 23 |
A |
913 |
902 |
3.79 |
|
|
|
| 24 |
A |
906 |
895 |
12.93 |
|
|
|
| 25 |
A |
743 |
734 |
5.22 |
|
|
|
| 26 |
A |
725 |
716 |
2.79 |
|
|
|
| 27 |
A |
573 |
566 |
5.95 |
|
|
|
| 28 |
A |
458 |
453 |
0.02 |
|
|
|
| 29 |
A |
391 |
386 |
3.67 |
|
|
|
| 30 |
A |
239 |
236 |
5.20 |
|
|
|
| 31 |
A |
188 |
185 |
0.16 |
|
|
|
| 32 |
A |
95 |
94 |
0.05 |
|
|
|
| 33 |
A |
13i |
13i |
0.09 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23855.3 cm
-1
Scaled (by 0.9879) Zero Point Vibrational Energy (zpe) 23566.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/Def2TZVPP
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-1.890 |
-0.504 |
0.000 |
| C2 |
-0.529 |
0.166 |
-0.000 |
| C3 |
0.684 |
-0.754 |
-0.000 |
| C4 |
2.023 |
-0.021 |
0.000 |
| O5 |
-0.417 |
1.382 |
-0.000 |
| H6 |
-2.681 |
0.253 |
-0.000 |
| H7 |
-1.996 |
-1.152 |
-0.883 |
| H8 |
-1.996 |
-1.151 |
0.884 |
| H9 |
2.859 |
-0.733 |
0.000 |
| H10 |
2.116 |
0.624 |
-0.882 |
| H11 |
2.116 |
0.624 |
0.883 |
| H12 |
0.594 |
-1.422 |
0.874 |
| H13 |
0.594 |
-1.422 |
-0.874 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
O5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.5169 | 2.5863 | 3.9425 | 2.3935 | 1.0952 | 1.1000 | 1.1000 | 4.7543 | 4.2540 | 4.2539 | 2.7883 | 2.7886 |
C2 | 1.5169 | | 1.5232 | 2.5591 | 1.2211 | 2.1537 | 2.1608 | 2.1607 | 3.5056 | 2.8256 | 2.8258 | 2.1327 | 2.1327 | C3 | 2.5863 | 1.5232 | | 1.5261 | 2.4035 | 3.5131 | 2.8503 | 2.8504 | 2.1747 | 2.1740 | 2.1740 | 1.1036 | 1.1036 | C4 | 3.9425 | 2.5591 | 1.5261 | | 2.8145 | 4.7122 | 4.2677 | 4.2676 | 1.0981 | 1.0970 | 1.0970 | 2.1837 | 2.1837 | O5 | 2.3935 | 1.2211 | 2.4035 | 2.8145 | | 2.5306 | 3.1139 | 3.1133 | 3.8993 | 2.7871 | 2.7872 | 3.1063 | 3.1064 | H6 | 1.0952 | 2.1537 | 3.5131 | 4.7122 | 2.5306 | | 1.7946 | 1.7946 | 5.6273 | 4.8916 | 4.8917 | 3.7809 | 3.7811 | H7 | 1.1000 | 2.1608 | 2.8503 | 4.2677 | 3.1139 | 1.7946 | | 1.7663 | 4.9528 | 4.4792 | 4.8145 | 3.1413 | 2.6046 | H8 | 1.1000 | 2.1607 | 2.8504 | 4.2676 | 3.1133 | 1.7946 | 1.7663 | | 4.9528 | 4.8144 | 4.4787 | 2.6045 | 3.1421 | H9 | 4.7543 | 3.5056 | 2.1747 | 1.0981 | 3.8993 | 5.6273 | 4.9528 | 4.9528 | | 1.7810 | 1.7810 | 2.5237 | 2.5236 | H10 | 4.2540 | 2.8256 | 2.1740 | 1.0970 | 2.7871 | 4.8916 | 4.4792 | 4.8144 | 1.7810 | | 1.7652 | 3.0962 | 2.5498 | H11 | 4.2539 | 2.8258 | 2.1740 | 1.0970 | 2.7872 | 4.8917 | 4.8145 | 4.4787 | 1.7810 | 1.7652 | | 2.5497 | 3.0962 | H12 | 2.7883 | 2.1327 | 1.1036 | 2.1837 | 3.1063 | 3.7809 | 3.1413 | 2.6045 | 2.5237 | 3.0962 | 2.5497 | | 1.7476 | H13 | 2.7886 | 2.1327 | 1.1036 | 2.1837 | 3.1064 | 3.7811 | 2.6046 | 3.1421 | 2.5236 | 2.5498 | 3.0962 | 1.7476 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
116.584 |
|
C1 |
C2 |
O5 |
121.526 |
| C2 |
C1 |
H6 |
110.033 |
|
C2 |
C1 |
H7 |
110.305 |
| C2 |
C1 |
H8 |
110.300 |
|
C2 |
C3 |
C4 |
114.115 |
| C2 |
C3 |
H12 |
107.486 |
|
C2 |
C3 |
H13 |
107.486 |
| C3 |
C2 |
O5 |
121.890 |
|
C3 |
C4 |
H9 |
110.875 |
| C3 |
C4 |
H10 |
110.889 |
|
C3 |
C4 |
H11 |
110.890 |
| C4 |
C3 |
H12 |
111.260 |
|
C4 |
C3 |
H13 |
111.260 |
| H6 |
C1 |
H7 |
109.671 |
|
H6 |
C1 |
H8 |
109.667 |
| H7 |
C1 |
H8 |
106.811 |
|
H9 |
C4 |
H10 |
108.462 |
| H9 |
C4 |
H11 |
108.461 |
|
H10 |
C4 |
H11 |
107.137 |
| H12 |
C3 |
H13 |
104.707 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.229 |
|
|
|
| 2 |
C |
0.148 |
|
|
|
| 3 |
C |
-0.091 |
|
|
|
| 4 |
C |
-0.241 |
|
|
|
| 5 |
O |
-0.258 |
|
|
|
| 6 |
H |
0.101 |
|
|
|
| 7 |
H |
0.087 |
|
|
|
| 8 |
H |
0.087 |
|
|
|
| 9 |
H |
0.080 |
|
|
|
| 10 |
H |
0.087 |
|
|
|
| 11 |
H |
0.087 |
|
|
|
| 12 |
H |
0.071 |
|
|
|
| 13 |
H |
0.071 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.180 |
-2.768 |
0.000 |
2.773 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.425 |
1.137 |
-0.000 |
| y |
1.137 |
-35.436 |
0.001 |
| z |
-0.000 |
0.001 |
-30.916 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
2.752 |
1.137 |
-0.000 |
| y |
1.137 |
-4.766 |
0.001 |
| z |
-0.000 |
0.001 |
2.015 |
|
| Polar |
|---|
| 3z2-r2 | 4.029 |
|---|
| x2-y2 | 5.012 |
|---|
| xy | 1.137 |
|---|
| xz | -0.000 |
|---|
| yz | 0.001 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.366 |
0.230 |
-0.000 |
| y |
0.230 |
8.339 |
-0.000 |
| z |
-0.000 |
-0.000 |
6.477 |
<r2> (average value of r
2) Å
2
| <r2> |
137.966 |
| (<r2>)1/2 |
11.746 |