Jump to
S1C2
Energy calculated at PBEPBE/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -342.170385 |
| Energy at 298.15K | |
| HF Energy | -342.170385 |
| Nuclear repulsion energy | 244.351421 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
2996 |
2959 |
48.25 |
|
|
|
| 2 |
A1 |
1842 |
1820 |
570.63 |
|
|
|
| 3 |
A1 |
1485 |
1467 |
0.10 |
|
|
|
| 4 |
A1 |
1334 |
1318 |
0.17 |
|
|
|
| 5 |
A1 |
1065 |
1052 |
191.87 |
|
|
|
| 6 |
A1 |
950 |
938 |
24.14 |
|
|
|
| 7 |
A1 |
860 |
850 |
3.57 |
|
|
|
| 8 |
A1 |
701 |
693 |
3.08 |
|
|
|
| 9 |
A2 |
3026 |
2989 |
0.00 |
|
|
|
| 10 |
A2 |
1202 |
1187 |
0.00 |
|
|
|
| 11 |
A2 |
1115 |
1101 |
0.00 |
|
|
|
| 12 |
A2 |
114i |
112i |
0.00 |
|
|
|
| 13 |
B1 |
3049 |
3012 |
29.44 |
|
|
|
| 14 |
B1 |
1199 |
1185 |
1.05 |
|
|
|
| 15 |
B1 |
829 |
819 |
0.14 |
|
|
|
| 16 |
B1 |
730 |
722 |
14.73 |
|
|
|
| 17 |
B1 |
183 |
180 |
1.29 |
|
|
|
| 18 |
B2 |
2991 |
2954 |
21.08 |
|
|
|
| 19 |
B2 |
1474 |
1456 |
6.26 |
|
|
|
| 20 |
B2 |
1360 |
1344 |
32.06 |
|
|
|
| 21 |
B2 |
1091 |
1077 |
251.12 |
|
|
|
| 22 |
B2 |
1029 |
1016 |
11.64 |
|
|
|
| 23 |
B2 |
723 |
715 |
0.00 |
|
|
|
| 24 |
B2 |
502 |
496 |
0.07 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 15809.6 cm
-1
Scaled (by 0.9879) Zero Point Vibrational Energy (zpe) 15618.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/Def2TZVPP
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.868 |
| O2 |
0.000 |
0.000 |
2.056 |
| O3 |
0.000 |
1.119 |
0.070 |
| O4 |
0.000 |
-1.119 |
0.070 |
| C5 |
0.000 |
0.778 |
-1.300 |
| C6 |
0.000 |
-0.778 |
-1.300 |
| H7 |
-0.891 |
1.193 |
-1.793 |
| H8 |
0.891 |
1.193 |
-1.793 |
| H9 |
0.891 |
-1.193 |
-1.793 |
| H10 |
-0.891 |
-1.193 |
-1.793 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.1878 | 1.3747 | 1.3747 | 2.3038 | 2.3038 | 3.0491 | 3.0491 | 3.0491 | 3.0491 |
O2 | 1.1878 | | 2.2794 | 2.2794 | 3.4452 | 3.4452 | 4.1265 | 4.1265 | 4.1265 | 4.1265 | O3 | 1.3747 | 2.2794 | | 2.2389 | 1.4124 | 2.3407 | 2.0665 | 2.0665 | 3.1001 | 3.1001 | O4 | 1.3747 | 2.2794 | 2.2389 | | 2.3407 | 1.4124 | 3.1001 | 3.1001 | 2.0665 | 2.0665 | C5 | 2.3038 | 3.4452 | 1.4124 | 2.3407 | | 1.5561 | 1.0996 | 1.0996 | 2.2183 | 2.2183 | C6 | 2.3038 | 3.4452 | 2.3407 | 1.4124 | 1.5561 | | 2.2183 | 2.2183 | 1.0996 | 1.0996 | H7 | 3.0491 | 4.1265 | 2.0665 | 3.1001 | 1.0996 | 2.2183 | | 1.7830 | 2.9780 | 2.3852 | H8 | 3.0491 | 4.1265 | 2.0665 | 3.1001 | 1.0996 | 2.2183 | 1.7830 | | 2.3852 | 2.9780 | H9 | 3.0491 | 4.1265 | 3.1001 | 2.0665 | 2.2183 | 1.0996 | 2.9780 | 2.3852 | | 1.7830 | H10 | 3.0491 | 4.1265 | 3.1001 | 2.0665 | 2.2183 | 1.0996 | 2.3852 | 2.9780 | 1.7830 | |
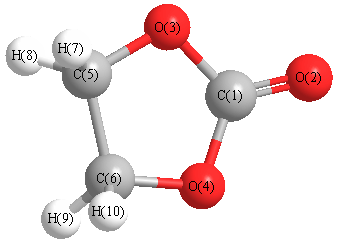 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
111.492 |
|
C1 |
O4 |
C6 |
111.492 |
| O2 |
C1 |
O3 |
125.479 |
|
O2 |
C1 |
O4 |
125.479 |
| O3 |
C1 |
O4 |
109.042 |
|
O3 |
C5 |
C6 |
103.987 |
| O3 |
C5 |
H7 |
110.082 |
|
O3 |
C5 |
H8 |
110.082 |
| O4 |
C6 |
C5 |
103.987 |
|
O4 |
C6 |
H9 |
110.082 |
| O4 |
C6 |
H10 |
110.082 |
|
C5 |
C6 |
H9 |
112.150 |
| C5 |
C6 |
H10 |
112.150 |
|
C6 |
C5 |
H7 |
112.150 |
| C6 |
C5 |
H8 |
112.150 |
|
H7 |
C5 |
H8 |
108.343 |
| H9 |
C6 |
H10 |
108.343 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.253 |
|
|
|
| 2 |
O |
-0.240 |
|
|
|
| 3 |
O |
-0.181 |
|
|
|
| 4 |
O |
-0.181 |
|
|
|
| 5 |
C |
-0.007 |
|
|
|
| 6 |
C |
-0.007 |
|
|
|
| 7 |
H |
0.091 |
|
|
|
| 8 |
H |
0.091 |
|
|
|
| 9 |
H |
0.091 |
|
|
|
| 10 |
H |
0.091 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-5.211 |
5.211 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.329 |
0.000 |
0.000 |
| y |
0.000 |
-36.017 |
0.000 |
| z |
0.000 |
0.000 |
-36.408 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.884 |
0.000 |
0.000 |
| y |
0.000 |
-1.648 |
0.000 |
| z |
0.000 |
0.000 |
-2.236 |
|
| Polar |
|---|
| 3z2-r2 | -4.471 |
|---|
| x2-y2 | 3.688 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.021 |
0.000 |
0.000 |
| y |
0.000 |
6.290 |
0.000 |
| z |
0.000 |
0.000 |
8.592 |
<r2> (average value of r
2) Å
2
| <r2> |
129.863 |
| (<r2>)1/2 |
11.396 |
Jump to
S1C1
Energy calculated at PBEPBE/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -342.170949 |
| Energy at 298.15K | -342.177316 |
| HF Energy | -342.170949 |
| Nuclear repulsion energy | 244.852982 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3050 |
3013 |
12.49 |
|
|
|
| 2 |
A |
2979 |
2943 |
28.70 |
|
|
|
| 3 |
A |
1845 |
1823 |
558.92 |
|
|
|
| 4 |
A |
1473 |
1455 |
0.14 |
|
|
|
| 5 |
A |
1337 |
1321 |
0.50 |
|
|
|
| 6 |
A |
1206 |
1192 |
11.61 |
|
|
|
| 7 |
A |
1114 |
1101 |
3.52 |
|
|
|
| 8 |
A |
1053 |
1040 |
175.26 |
|
|
|
| 9 |
A |
946 |
934 |
22.38 |
|
|
|
| 10 |
A |
860 |
850 |
6.05 |
|
|
|
| 11 |
A |
699 |
690 |
3.23 |
|
|
|
| 12 |
A |
155 |
153 |
0.25 |
|
|
|
| 13 |
B |
3062 |
3025 |
19.02 |
|
|
|
| 14 |
B |
2985 |
2949 |
34.30 |
|
|
|
| 15 |
B |
1465 |
1447 |
7.60 |
|
|
|
| 16 |
B |
1351 |
1335 |
26.94 |
|
|
|
| 17 |
B |
1197 |
1183 |
5.38 |
|
|
|
| 18 |
B |
1077 |
1064 |
243.54 |
|
|
|
| 19 |
B |
1019 |
1007 |
6.04 |
|
|
|
| 20 |
B |
860 |
849 |
0.60 |
|
|
|
| 21 |
B |
737 |
728 |
14.76 |
|
|
|
| 22 |
B |
667 |
659 |
0.34 |
|
|
|
| 23 |
B |
502 |
496 |
0.08 |
|
|
|
| 24 |
B |
185 |
183 |
1.13 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 15911.9 cm
-1
Scaled (by 0.9879) Zero Point Vibrational Energy (zpe) 15719.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/Def2TZVPP
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.861 |
| O2 |
0.000 |
0.000 |
2.053 |
| O3 |
0.000 |
1.128 |
0.060 |
| O4 |
0.000 |
-1.128 |
0.060 |
| C5 |
0.223 |
0.731 |
-1.295 |
| C6 |
-0.223 |
-0.731 |
-1.295 |
| H7 |
-0.365 |
1.369 |
-1.966 |
| H8 |
1.294 |
0.831 |
-1.535 |
| H9 |
0.365 |
-1.369 |
-1.966 |
| H10 |
-1.294 |
-0.831 |
-1.535 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.1916 | 1.3837 | 1.3837 | 2.2873 | 2.2873 | 3.1619 | 2.8472 | 3.1619 | 2.8472 |
O2 | 1.1916 | | 2.2901 | 2.2901 | 3.4335 | 3.4335 | 4.2608 | 3.9034 | 4.2608 | 3.9034 | O3 | 1.3837 | 2.2901 | | 2.2562 | 1.4288 | 2.3111 | 2.0721 | 2.0749 | 3.2358 | 2.8384 | O4 | 1.3837 | 2.2901 | 2.2562 | | 2.3111 | 1.4288 | 3.2358 | 2.8384 | 2.0721 | 2.0749 | C5 | 2.2873 | 3.4335 | 1.4288 | 2.3111 | | 1.5289 | 1.0964 | 1.1022 | 2.2092 | 2.1910 | C6 | 2.2873 | 3.4335 | 2.3111 | 1.4288 | 1.5289 | | 2.2092 | 2.1910 | 1.0964 | 1.1022 | H7 | 3.1619 | 4.2608 | 2.0721 | 3.2358 | 1.0964 | 2.2092 | | 1.7959 | 2.8330 | 2.4269 | H8 | 2.8472 | 3.9034 | 2.0749 | 2.8384 | 1.1022 | 2.1910 | 1.7959 | | 2.4269 | 3.0760 | H9 | 3.1619 | 4.2608 | 3.2358 | 2.0721 | 2.2092 | 1.0964 | 2.8330 | 2.4269 | | 1.7959 | H10 | 2.8472 | 3.9034 | 2.8384 | 2.0749 | 2.1910 | 1.1022 | 2.4269 | 3.0760 | 1.7959 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
108.815 |
|
C1 |
O4 |
C6 |
108.815 |
| O2 |
C1 |
O3 |
125.389 |
|
O2 |
C1 |
O4 |
125.389 |
| O3 |
C1 |
O4 |
109.222 |
|
O3 |
C5 |
C6 |
102.719 |
| O3 |
C5 |
H7 |
109.580 |
|
O3 |
C5 |
H8 |
109.455 |
| O4 |
C6 |
C5 |
102.719 |
|
O4 |
C6 |
H9 |
109.580 |
| O4 |
C6 |
H10 |
109.455 |
|
C5 |
C6 |
H9 |
113.572 |
| C5 |
C6 |
H10 |
111.734 |
|
C6 |
C5 |
H7 |
113.572 |
| C6 |
C5 |
H8 |
111.734 |
|
H7 |
C5 |
H8 |
109.537 |
| H9 |
C6 |
H10 |
109.537 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.252 |
|
|
|
| 2 |
O |
-0.236 |
|
|
|
| 3 |
O |
-0.185 |
|
|
|
| 4 |
O |
-0.185 |
|
|
|
| 5 |
C |
-0.008 |
|
|
|
| 6 |
C |
-0.008 |
|
|
|
| 7 |
H |
0.095 |
|
|
|
| 8 |
H |
0.089 |
|
|
|
| 9 |
H |
0.095 |
|
|
|
| 10 |
H |
0.089 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-5.169 |
5.169 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.395 |
0.244 |
0.000 |
| y |
0.244 |
-36.056 |
0.000 |
| z |
0.000 |
0.000 |
-36.233 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.750 |
0.244 |
0.000 |
| y |
0.244 |
-1.742 |
0.000 |
| z |
0.000 |
0.000 |
-2.008 |
|
| Polar |
|---|
| 3z2-r2 | -4.016 |
|---|
| x2-y2 | 3.661 |
|---|
| xy | 0.244 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.124 |
0.174 |
0.000 |
| y |
0.174 |
6.236 |
0.000 |
| z |
0.000 |
0.000 |
8.560 |
<r2> (average value of r
2) Å
2
| <r2> |
128.897 |
| (<r2>)1/2 |
11.353 |