Vibrational Frequencies calculated at PBEPBE/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3570 |
3529 |
73.73 |
|
|
|
| 2 |
A' |
3191 |
3154 |
0.41 |
|
|
|
| 3 |
A' |
3127 |
3091 |
11.60 |
|
|
|
| 4 |
A' |
3111 |
3075 |
12.71 |
|
|
|
| 5 |
A' |
1605 |
1586 |
57.19 |
|
|
|
| 6 |
A' |
1568 |
1550 |
68.26 |
|
|
|
| 7 |
A' |
1475 |
1459 |
26.29 |
|
|
|
| 8 |
A' |
1440 |
1424 |
1.50 |
|
|
|
| 9 |
A' |
1392 |
1376 |
50.81 |
|
|
|
| 10 |
A' |
1374 |
1359 |
17.24 |
|
|
|
| 11 |
A' |
1331 |
1316 |
54.31 |
|
|
|
| 12 |
A' |
1294 |
1279 |
23.94 |
|
|
|
| 13 |
A' |
1276 |
1262 |
19.50 |
|
|
|
| 14 |
A' |
1246 |
1232 |
24.76 |
|
|
|
| 15 |
A' |
1170 |
1156 |
5.22 |
|
|
|
| 16 |
A' |
1103 |
1090 |
7.25 |
|
|
|
| 17 |
A' |
1057 |
1044 |
14.65 |
|
|
|
| 18 |
A' |
914 |
903 |
0.48 |
|
|
|
| 19 |
A' |
882 |
872 |
9.82 |
|
|
|
| 20 |
A' |
784 |
775 |
10.68 |
|
|
|
| 21 |
A' |
638 |
631 |
0.23 |
|
|
|
| 22 |
A' |
551 |
545 |
3.09 |
|
|
|
| 23 |
A' |
432 |
427 |
12.51 |
|
|
|
| 24 |
A" |
946 |
935 |
0.07 |
|
|
|
| 25 |
A" |
895 |
884 |
13.53 |
|
|
|
| 26 |
A" |
828 |
819 |
10.66 |
|
|
|
| 27 |
A" |
768 |
759 |
8.78 |
|
|
|
| 28 |
A" |
641 |
634 |
3.20 |
|
|
|
| 29 |
A" |
600 |
593 |
26.39 |
|
|
|
| 30 |
A" |
513 |
507 |
88.18 |
|
|
|
| 31 |
A" |
394 |
390 |
2.05 |
|
|
|
| 32 |
A" |
230 |
228 |
0.28 |
|
|
|
| 33 |
A" |
219 |
217 |
4.64 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20281.0 cm
-1
Scaled (by 0.9886) Zero Point Vibrational Energy (zpe) 20049.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
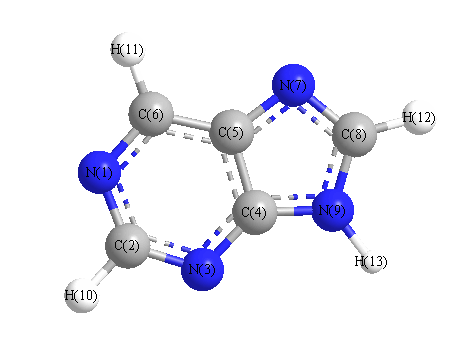
Charges from optimized geometry at PBEPBE/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.180 |
|
|
|
| 2 |
C |
0.026 |
|
|
|
| 3 |
N |
-0.248 |
|
|
|
| 4 |
C |
0.045 |
|
|
|
| 5 |
C |
0.400 |
|
|
|
| 6 |
C |
-0.342 |
|
|
|
| 7 |
N |
-0.325 |
|
|
|
| 8 |
C |
0.193 |
|
|
|
| 9 |
N |
-0.362 |
|
|
|
| 10 |
H |
0.149 |
|
|
|
| 11 |
H |
0.164 |
|
|
|
| 12 |
H |
0.164 |
|
|
|
| 13 |
H |
0.315 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.270 |
3.022 |
0.000 |
3.779 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-51.453 |
6.337 |
0.000 |
| y |
6.337 |
-48.345 |
0.000 |
| z |
0.000 |
0.000 |
-52.705 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.929 |
6.337 |
0.000 |
| y |
6.337 |
3.734 |
0.000 |
| z |
0.000 |
0.000 |
-2.806 |
|
| Polar |
|---|
| 3z2-r2 | -5.611 |
|---|
| x2-y2 | -3.109 |
|---|
| xy | 6.337 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
17.188 |
1.388 |
0.000 |
| y |
1.388 |
13.622 |
0.000 |
| z |
0.000 |
0.000 |
6.682 |
<r2> (average value of r
2) Å
2
| <r2> |
257.736 |
| (<r2>)1/2 |
16.054 |