Jump to
S1C2
Energy calculated at PBEPBE/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -311.972577 |
| Energy at 298.15K | -311.987875 |
| HF Energy | -311.972577 |
| Nuclear repulsion energy | 313.580191 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3040 |
3019 |
39.49 |
|
|
|
| 2 |
A1 |
2971 |
2951 |
61.85 |
|
|
|
| 3 |
A1 |
2964 |
2943 |
6.30 |
|
|
|
| 4 |
A1 |
2871 |
2852 |
97.90 |
|
|
|
| 5 |
A1 |
1478 |
1468 |
3.19 |
|
|
|
| 6 |
A1 |
1456 |
1446 |
6.35 |
|
|
|
| 7 |
A1 |
1442 |
1432 |
0.05 |
|
|
|
| 8 |
A1 |
1395 |
1385 |
0.75 |
|
|
|
| 9 |
A1 |
1359 |
1350 |
2.73 |
|
|
|
| 10 |
A1 |
1290 |
1281 |
1.50 |
|
|
|
| 11 |
A1 |
1138 |
1130 |
1.49 |
|
|
|
| 12 |
A1 |
1039 |
1032 |
5.88 |
|
|
|
| 13 |
A1 |
974 |
968 |
17.75 |
|
|
|
| 14 |
A1 |
897 |
891 |
8.08 |
|
|
|
| 15 |
A1 |
418 |
415 |
0.39 |
|
|
|
| 16 |
A1 |
310 |
308 |
0.25 |
|
|
|
| 17 |
A1 |
100 |
99 |
0.14 |
|
|
|
| 18 |
A2 |
3030 |
3009 |
0.00 |
|
|
|
| 19 |
A2 |
3001 |
2981 |
0.00 |
|
|
|
| 20 |
A2 |
2889 |
2870 |
0.00 |
|
|
|
| 21 |
A2 |
1448 |
1438 |
0.00 |
|
|
|
| 22 |
A2 |
1270 |
1262 |
0.00 |
|
|
|
| 23 |
A2 |
1221 |
1212 |
0.00 |
|
|
|
| 24 |
A2 |
1134 |
1126 |
0.00 |
|
|
|
| 25 |
A2 |
868 |
862 |
0.00 |
|
|
|
| 26 |
A2 |
742 |
737 |
0.00 |
|
|
|
| 27 |
A2 |
222 |
220 |
0.00 |
|
|
|
| 28 |
A2 |
137 |
136 |
0.00 |
|
|
|
| 29 |
A2 |
77 |
76 |
0.00 |
|
|
|
| 30 |
B1 |
3030 |
3009 |
106.97 |
|
|
|
| 31 |
B1 |
3001 |
2981 |
8.81 |
|
|
|
| 32 |
B1 |
2884 |
2864 |
112.05 |
|
|
|
| 33 |
B1 |
1448 |
1438 |
14.71 |
|
|
|
| 34 |
B1 |
1272 |
1263 |
0.72 |
|
|
|
| 35 |
B1 |
1226 |
1218 |
2.96 |
|
|
|
| 36 |
B1 |
1157 |
1149 |
3.91 |
|
|
|
| 37 |
B1 |
880 |
874 |
2.83 |
|
|
|
| 38 |
B1 |
743 |
738 |
4.50 |
|
|
|
| 39 |
B1 |
222 |
220 |
0.21 |
|
|
|
| 40 |
B1 |
150 |
149 |
4.17 |
|
|
|
| 41 |
B1 |
56 |
55 |
0.20 |
|
|
|
| 42 |
B2 |
3040 |
3019 |
21.16 |
|
|
|
| 43 |
B2 |
2971 |
2950 |
8.38 |
|
|
|
| 44 |
B2 |
2963 |
2943 |
33.38 |
|
|
|
| 45 |
B2 |
2860 |
2840 |
18.16 |
|
|
|
| 46 |
B2 |
1464 |
1454 |
7.88 |
|
|
|
| 47 |
B2 |
1452 |
1442 |
0.01 |
|
|
|
| 48 |
B2 |
1441 |
1431 |
3.13 |
|
|
|
| 49 |
B2 |
1360 |
1351 |
8.89 |
|
|
|
| 50 |
B2 |
1346 |
1337 |
20.16 |
|
|
|
| 51 |
B2 |
1277 |
1268 |
8.44 |
|
|
|
| 52 |
B2 |
1109 |
1101 |
248.52 |
|
|
|
| 53 |
B2 |
1092 |
1085 |
2.18 |
|
|
|
| 54 |
B2 |
1030 |
1022 |
0.13 |
|
|
|
| 55 |
B2 |
875 |
869 |
1.91 |
|
|
|
| 56 |
B2 |
500 |
497 |
4.90 |
|
|
|
| 57 |
B2 |
241 |
239 |
0.51 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 41134.9 cm
-1
Scaled (by 0.9931) Zero Point Vibrational Energy (zpe) 40851.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/cc-pVTZ
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.404 |
| C2 |
0.000 |
1.182 |
-0.385 |
| C3 |
0.000 |
-1.182 |
-0.385 |
| C4 |
0.000 |
2.394 |
0.534 |
| C5 |
0.000 |
-2.394 |
0.534 |
| C6 |
0.000 |
3.711 |
-0.243 |
| C7 |
0.000 |
-3.711 |
-0.243 |
| H8 |
0.892 |
1.200 |
-1.046 |
| H9 |
-0.892 |
1.200 |
-1.046 |
| H10 |
-0.892 |
-1.200 |
-1.046 |
| H11 |
0.892 |
-1.200 |
-1.046 |
| H12 |
0.882 |
2.336 |
1.189 |
| H13 |
-0.882 |
2.336 |
1.189 |
| H14 |
-0.882 |
-2.336 |
1.189 |
| H15 |
0.882 |
-2.336 |
1.189 |
| H16 |
0.000 |
4.572 |
0.438 |
| H17 |
-0.887 |
3.797 |
-0.887 |
| H18 |
0.887 |
3.797 |
-0.887 |
| H19 |
0.000 |
-4.572 |
0.438 |
| H20 |
0.887 |
-3.797 |
-0.887 |
| H21 |
-0.887 |
-3.797 |
-0.887 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
C4 |
C5 |
C6 |
C7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
H21 |
| O1 | | 1.4209 | 1.4209 | 2.3974 | 2.3974 | 3.7666 | 3.7666 | 2.0827 | 2.0827 | 2.0827 | 2.0827 | 2.6179 | 2.6179 | 2.6179 | 2.6179 | 4.5726 | 4.1076 | 4.1076 | 4.5726 | 4.1076 | 4.1076 |
C2 | 1.4209 | | 2.3639 | 1.5206 | 3.6919 | 2.5326 | 4.8946 | 1.1102 | 1.1102 | 2.6280 | 2.6280 | 2.1422 | 2.1422 | 3.9540 | 3.9540 | 3.4888 | 2.8069 | 2.8069 | 5.8129 | 5.0823 | 5.0823 | C3 | 1.4209 | 2.3639 | | 3.6919 | 1.5206 | 4.8946 | 2.5326 | 2.6280 | 2.6280 | 1.1102 | 1.1102 | 3.9540 | 3.9540 | 2.1422 | 2.1422 | 5.8129 | 5.0823 | 5.0823 | 3.4888 | 2.8069 | 2.8069 | C4 | 2.3974 | 1.5206 | 3.6919 | | 4.7878 | 1.5287 | 6.1537 | 2.1715 | 2.1715 | 4.0258 | 4.0258 | 1.1008 | 1.1008 | 4.8562 | 4.8562 | 2.1807 | 2.1854 | 2.1854 | 6.9670 | 6.4136 | 6.4136 | C5 | 2.3974 | 3.6919 | 1.5206 | 4.7878 | | 6.1537 | 1.5287 | 4.0258 | 4.0258 | 2.1715 | 2.1715 | 4.8562 | 4.8562 | 1.1008 | 1.1008 | 6.9670 | 6.4136 | 6.4136 | 2.1807 | 2.1854 | 2.1854 | C6 | 3.7666 | 2.5326 | 4.8946 | 1.5287 | 6.1537 | | 7.4212 | 2.7825 | 2.7825 | 5.0552 | 5.0552 | 2.1723 | 2.1723 | 6.2765 | 6.2765 | 1.0982 | 1.1001 | 1.1001 | 8.3110 | 7.5873 | 7.5873 | C7 | 3.7666 | 4.8946 | 2.5326 | 6.1537 | 1.5287 | 7.4212 | | 5.0552 | 5.0552 | 2.7825 | 2.7825 | 6.2765 | 6.2765 | 2.1723 | 2.1723 | 8.3110 | 7.5873 | 7.5873 | 1.0982 | 1.1001 | 1.1001 | H8 | 2.0827 | 1.1102 | 2.6280 | 2.1715 | 4.0258 | 2.7825 | 5.0552 | | 1.7832 | 2.9902 | 2.4003 | 2.5076 | 3.0716 | 4.5442 | 4.1837 | 3.7906 | 3.1518 | 2.6017 | 6.0265 | 4.9996 | 5.3067 | H9 | 2.0827 | 1.1102 | 2.6280 | 2.1715 | 4.0258 | 2.7825 | 5.0552 | 1.7832 | | 2.4003 | 2.9902 | 3.0716 | 2.5076 | 4.1837 | 4.5442 | 3.7906 | 2.6017 | 3.1518 | 6.0265 | 5.3067 | 4.9996 | H10 | 2.0827 | 2.6280 | 1.1102 | 4.0258 | 2.1715 | 5.0552 | 2.7825 | 2.9902 | 2.4003 | | 1.7832 | 4.5442 | 4.1837 | 2.5076 | 3.0716 | 6.0265 | 4.9996 | 5.3067 | 3.7906 | 3.1518 | 2.6017 | H11 | 2.0827 | 2.6280 | 1.1102 | 4.0258 | 2.1715 | 5.0552 | 2.7825 | 2.4003 | 2.9902 | 1.7832 | | 4.1837 | 4.5442 | 3.0716 | 2.5076 | 6.0265 | 5.3067 | 4.9996 | 3.7906 | 2.6017 | 3.1518 | H12 | 2.6179 | 2.1422 | 3.9540 | 1.1008 | 4.8562 | 2.1723 | 6.2765 | 2.5076 | 3.0716 | 4.5442 | 4.1837 | | 1.7646 | 4.9946 | 4.6725 | 2.5188 | 3.0950 | 2.5391 | 7.0053 | 6.4753 | 6.7128 | H13 | 2.6179 | 2.1422 | 3.9540 | 1.1008 | 4.8562 | 2.1723 | 6.2765 | 3.0716 | 2.5076 | 4.1837 | 4.5442 | 1.7646 | | 4.6725 | 4.9946 | 2.5188 | 2.5391 | 3.0950 | 7.0053 | 6.7128 | 6.4753 | H14 | 2.6179 | 3.9540 | 2.1422 | 4.8562 | 1.1008 | 6.2765 | 2.1723 | 4.5442 | 4.1837 | 2.5076 | 3.0716 | 4.9946 | 4.6725 | | 1.7646 | 7.0053 | 6.4753 | 6.7128 | 2.5188 | 3.0950 | 2.5391 | H15 | 2.6179 | 3.9540 | 2.1422 | 4.8562 | 1.1008 | 6.2765 | 2.1723 | 4.1837 | 4.5442 | 3.0716 | 2.5076 | 4.6725 | 4.9946 | 1.7646 | | 7.0053 | 6.7128 | 6.4753 | 2.5188 | 2.5391 | 3.0950 | H16 | 4.5726 | 3.4888 | 5.8129 | 2.1807 | 6.9670 | 1.0982 | 8.3110 | 3.7906 | 3.7906 | 6.0265 | 6.0265 | 2.5188 | 2.5188 | 7.0053 | 7.0053 | | 1.7733 | 1.7733 | 9.1450 | 8.5201 | 8.5201 | H17 | 4.1076 | 2.8069 | 5.0823 | 2.1854 | 6.4136 | 1.1001 | 7.5873 | 3.1518 | 2.6017 | 4.9996 | 5.3067 | 3.0950 | 2.5391 | 6.4753 | 6.7128 | 1.7733 | | 1.7748 | 8.5201 | 7.7986 | 7.5940 | H18 | 4.1076 | 2.8069 | 5.0823 | 2.1854 | 6.4136 | 1.1001 | 7.5873 | 2.6017 | 3.1518 | 5.3067 | 4.9996 | 2.5391 | 3.0950 | 6.7128 | 6.4753 | 1.7733 | 1.7748 | | 8.5201 | 7.5940 | 7.7986 | H19 | 4.5726 | 5.8129 | 3.4888 | 6.9670 | 2.1807 | 8.3110 | 1.0982 | 6.0265 | 6.0265 | 3.7906 | 3.7906 | 7.0053 | 7.0053 | 2.5188 | 2.5188 | 9.1450 | 8.5201 | 8.5201 | | 1.7733 | 1.7733 | H20 | 4.1076 | 5.0823 | 2.8069 | 6.4136 | 2.1854 | 7.5873 | 1.1001 | 4.9996 | 5.3067 | 3.1518 | 2.6017 | 6.4753 | 6.7128 | 3.0950 | 2.5391 | 8.5201 | 7.7986 | 7.5940 | 1.7733 | | 1.7748 | H21 | 4.1076 | 5.0823 | 2.8069 | 6.4136 | 2.1854 | 7.5873 | 1.1001 | 5.3067 | 4.9996 | 2.6017 | 3.1518 | 6.7128 | 6.4753 | 2.5391 | 3.0950 | 8.5201 | 7.5940 | 7.7986 | 1.7733 | 1.7748 | |
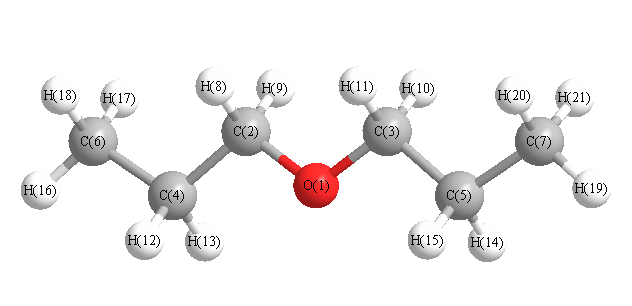 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C2 |
C4 |
109.130 |
|
O1 |
C2 |
H8 |
110.134 |
| O1 |
C2 |
H9 |
110.134 |
|
O1 |
C3 |
C5 |
109.130 |
| O1 |
C3 |
H10 |
110.134 |
|
O1 |
C3 |
H11 |
110.134 |
| C2 |
O1 |
C3 |
112.571 |
|
C2 |
C4 |
C6 |
112.309 |
| C2 |
C4 |
H12 |
108.545 |
|
C2 |
C4 |
H13 |
108.545 |
| C3 |
C5 |
C7 |
112.309 |
|
C3 |
C5 |
H14 |
108.545 |
| C3 |
C5 |
H15 |
108.545 |
|
C4 |
C2 |
H8 |
110.286 |
| C4 |
C2 |
H9 |
110.286 |
|
C4 |
C6 |
H16 |
111.171 |
| C4 |
C6 |
H17 |
111.425 |
|
C4 |
C6 |
H18 |
111.425 |
| C5 |
C3 |
H10 |
110.286 |
|
C5 |
C3 |
H11 |
110.286 |
| C5 |
C7 |
H19 |
111.171 |
|
C5 |
C7 |
H20 |
111.425 |
| C5 |
C7 |
H21 |
111.425 |
|
C6 |
C4 |
H12 |
110.350 |
| C6 |
C4 |
H13 |
110.350 |
|
C7 |
C5 |
H14 |
110.350 |
| C7 |
C5 |
H15 |
110.350 |
|
H8 |
C2 |
H9 |
106.853 |
| H10 |
C3 |
H11 |
106.853 |
|
H12 |
C4 |
H13 |
106.548 |
| H14 |
C5 |
H15 |
106.548 |
|
H16 |
C6 |
H17 |
107.539 |
| H16 |
C6 |
H18 |
107.539 |
|
H17 |
C6 |
H18 |
107.538 |
| H19 |
C7 |
H20 |
107.539 |
|
H19 |
C7 |
H21 |
107.539 |
| H20 |
C7 |
H21 |
107.538 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.248 |
|
|
|
| 2 |
C |
0.048 |
|
|
|
| 3 |
C |
0.048 |
|
|
|
| 4 |
C |
-0.150 |
|
|
|
| 5 |
C |
-0.150 |
|
|
|
| 6 |
C |
-0.303 |
|
|
|
| 7 |
C |
-0.303 |
|
|
|
| 8 |
H |
0.038 |
|
|
|
| 9 |
H |
0.038 |
|
|
|
| 10 |
H |
0.038 |
|
|
|
| 11 |
H |
0.038 |
|
|
|
| 12 |
H |
0.086 |
|
|
|
| 13 |
H |
0.086 |
|
|
|
| 14 |
H |
0.086 |
|
|
|
| 15 |
H |
0.086 |
|
|
|
| 16 |
H |
0.100 |
|
|
|
| 17 |
H |
0.090 |
|
|
|
| 18 |
H |
0.090 |
|
|
|
| 19 |
H |
0.100 |
|
|
|
| 20 |
H |
0.090 |
|
|
|
| 21 |
H |
0.090 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.787 |
0.787 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.562 |
0.000 |
0.000 |
| y |
0.000 |
-44.457 |
0.000 |
| z |
0.000 |
0.000 |
-47.945 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.361 |
0.000 |
0.000 |
| y |
0.000 |
2.796 |
0.000 |
| z |
0.000 |
0.000 |
-2.435 |
|
| Polar |
|---|
| 3z2-r2 | -4.871 |
|---|
| x2-y2 | -2.105 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.570 |
0.000 |
0.000 |
| y |
0.000 |
15.749 |
0.000 |
| z |
0.000 |
0.000 |
11.193 |
<r2> (average value of r
2) Å
2
| <r2> |
436.983 |
| (<r2>)1/2 |
20.904 |
Jump to
S1C1
Energy calculated at PBEPBE/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -311.973065 |
| Energy at 298.15K | -311.988559 |
| HF Energy | -311.973065 |
| Nuclear repulsion energy | 323.433606 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3051 |
3030 |
5.90 |
|
|
|
| 2 |
A |
3033 |
3012 |
54.37 |
|
|
|
| 3 |
A |
2997 |
2976 |
11.64 |
|
|
|
| 4 |
A |
2968 |
2947 |
9.16 |
|
|
|
| 5 |
A |
2964 |
2944 |
1.32 |
|
|
|
| 6 |
A |
2898 |
2878 |
16.09 |
|
|
|
| 7 |
A |
2872 |
2852 |
95.95 |
|
|
|
| 8 |
A |
1472 |
1461 |
4.23 |
|
|
|
| 9 |
A |
1458 |
1448 |
4.36 |
|
|
|
| 10 |
A |
1444 |
1434 |
5.47 |
|
|
|
| 11 |
A |
1425 |
1415 |
0.77 |
|
|
|
| 12 |
A |
1390 |
1380 |
4.45 |
|
|
|
| 13 |
A |
1355 |
1345 |
1.46 |
|
|
|
| 14 |
A |
1328 |
1318 |
0.37 |
|
|
|
| 15 |
A |
1271 |
1262 |
2.75 |
|
|
|
| 16 |
A |
1222 |
1214 |
0.07 |
|
|
|
| 17 |
A |
1138 |
1130 |
5.08 |
|
|
|
| 18 |
A |
1105 |
1097 |
7.23 |
|
|
|
| 19 |
A |
1056 |
1049 |
8.51 |
|
|
|
| 20 |
A |
923 |
917 |
6.55 |
|
|
|
| 21 |
A |
892 |
885 |
8.27 |
|
|
|
| 22 |
A |
880 |
874 |
0.17 |
|
|
|
| 23 |
A |
726 |
721 |
0.18 |
|
|
|
| 24 |
A |
467 |
463 |
0.11 |
|
|
|
| 25 |
A |
322 |
320 |
0.23 |
|
|
|
| 26 |
A |
257 |
256 |
0.56 |
|
|
|
| 27 |
A |
163 |
162 |
0.05 |
|
|
|
| 28 |
A |
108 |
107 |
0.02 |
|
|
|
| 29 |
A |
44 |
44 |
0.00 |
|
|
|
| 30 |
B |
3050 |
3029 |
46.34 |
|
|
|
| 31 |
B |
3033 |
3012 |
24.72 |
|
|
|
| 32 |
B |
2997 |
2976 |
24.50 |
|
|
|
| 33 |
B |
2968 |
2947 |
66.43 |
|
|
|
| 34 |
B |
2964 |
2944 |
44.91 |
|
|
|
| 35 |
B |
2892 |
2872 |
103.34 |
|
|
|
| 36 |
B |
2861 |
2842 |
23.87 |
|
|
|
| 37 |
B |
1457 |
1447 |
6.88 |
|
|
|
| 38 |
B |
1453 |
1443 |
4.22 |
|
|
|
| 39 |
B |
1442 |
1432 |
10.77 |
|
|
|
| 40 |
B |
1425 |
1415 |
5.29 |
|
|
|
| 41 |
B |
1358 |
1349 |
13.77 |
|
|
|
| 42 |
B |
1336 |
1327 |
16.01 |
|
|
|
| 43 |
B |
1322 |
1313 |
19.33 |
|
|
|
| 44 |
B |
1261 |
1252 |
2.20 |
|
|
|
| 45 |
B |
1229 |
1220 |
5.94 |
|
|
|
| 46 |
B |
1140 |
1132 |
4.61 |
|
|
|
| 47 |
B |
1113 |
1106 |
159.10 |
|
|
|
| 48 |
B |
1074 |
1067 |
26.43 |
|
|
|
| 49 |
B |
996 |
989 |
49.48 |
|
|
|
| 50 |
B |
903 |
897 |
0.86 |
|
|
|
| 51 |
B |
860 |
854 |
0.49 |
|
|
|
| 52 |
B |
760 |
755 |
2.60 |
|
|
|
| 53 |
B |
482 |
479 |
1.26 |
|
|
|
| 54 |
B |
314 |
312 |
0.62 |
|
|
|
| 55 |
B |
210 |
208 |
1.63 |
|
|
|
| 56 |
B |
160 |
158 |
3.28 |
|
|
|
| 57 |
B |
72 |
72 |
1.15 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 41179.5 cm
-1
Scaled (by 0.9931) Zero Point Vibrational Energy (zpe) 40895.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/cc-pVTZ
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.096 |
| C2 |
0.015 |
1.183 |
0.885 |
| C3 |
-0.015 |
-1.183 |
0.885 |
| C4 |
0.000 |
2.397 |
-0.032 |
| C5 |
0.000 |
-2.397 |
-0.032 |
| C6 |
-1.273 |
2.510 |
-0.871 |
| C7 |
1.273 |
-2.510 |
-0.871 |
| H8 |
-0.869 |
1.199 |
1.557 |
| H9 |
0.917 |
1.195 |
1.529 |
| H10 |
0.869 |
-1.199 |
1.557 |
| H11 |
-0.917 |
-1.195 |
1.529 |
| H12 |
0.883 |
2.348 |
-0.689 |
| H13 |
0.124 |
3.295 |
0.595 |
| H14 |
-0.883 |
-2.348 |
-0.689 |
| H15 |
-0.124 |
-3.295 |
0.595 |
| H16 |
-1.242 |
3.389 |
-1.529 |
| H17 |
-1.406 |
1.619 |
-1.499 |
| H18 |
-2.162 |
2.603 |
-0.230 |
| H19 |
1.242 |
-3.389 |
-1.529 |
| H20 |
1.406 |
-1.619 |
-1.499 |
| H21 |
2.162 |
-2.603 |
-0.230 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
C4 |
C5 |
C6 |
C7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
H21 |
| O1 | | 1.4221 | 1.4221 | 2.4005 | 2.4005 | 2.9757 | 2.9757 | 2.0804 | 2.0797 | 2.0804 | 2.0797 | 2.6281 | 3.3345 | 2.6281 | 3.3345 | 3.9582 | 2.6720 | 3.3999 | 3.9582 | 2.6720 | 3.3999 |
C2 | 1.4221 | | 2.3663 | 1.5215 | 3.6958 | 2.5500 | 4.2783 | 1.1106 | 1.1088 | 2.6182 | 2.6345 | 2.1415 | 2.1341 | 3.9684 | 4.4892 | 3.5031 | 2.8088 | 2.8282 | 5.3138 | 3.9328 | 4.4935 | C3 | 1.4221 | 2.3663 | | 3.6958 | 1.5215 | 4.2783 | 2.5500 | 2.6182 | 2.6345 | 1.1106 | 1.1088 | 3.9684 | 4.4892 | 2.1415 | 2.1341 | 5.3138 | 3.9328 | 4.4935 | 3.5031 | 2.8088 | 2.8282 | C4 | 2.4005 | 1.5215 | 3.6958 | | 4.7942 | 1.5291 | 5.1382 | 2.1721 | 2.1734 | 4.0263 | 4.0232 | 1.1014 | 1.1018 | 4.8708 | 5.7275 | 2.1833 | 2.1757 | 2.1812 | 6.1042 | 4.5005 | 5.4516 | C5 | 2.4005 | 3.6958 | 1.5215 | 4.7942 | | 5.1382 | 1.5291 | 4.0263 | 4.0232 | 2.1721 | 2.1734 | 4.8708 | 5.7275 | 1.1014 | 1.1018 | 6.1042 | 4.5005 | 5.4516 | 2.1833 | 2.1757 | 2.1812 | C6 | 2.9757 | 2.5500 | 4.2783 | 1.5291 | 5.1382 | | 5.6283 | 2.7892 | 3.5053 | 4.9233 | 4.4292 | 2.1704 | 2.1722 | 4.8764 | 6.0957 | 1.0987 | 1.0977 | 1.1005 | 6.4461 | 4.9614 | 6.1935 | C7 | 2.9757 | 4.2783 | 2.5500 | 5.1382 | 1.5291 | 5.6283 | | 4.9233 | 4.4292 | 2.7892 | 3.5053 | 4.8764 | 6.0957 | 2.1704 | 2.1722 | 6.4461 | 4.9614 | 6.1935 | 1.0987 | 1.0977 | 1.1005 | H8 | 2.0804 | 1.1106 | 2.6182 | 2.1721 | 4.0263 | 2.7892 | 4.9233 | | 1.7859 | 2.9611 | 2.3947 | 3.0715 | 2.5111 | 4.1977 | 4.6552 | 3.8029 | 3.1310 | 2.6150 | 5.9184 | 4.7384 | 5.1805 | H9 | 2.0797 | 1.1088 | 2.6345 | 2.1734 | 4.0232 | 3.5053 | 4.4292 | 1.7859 | | 2.3947 | 3.0128 | 2.4998 | 2.4308 | 4.5512 | 4.7029 | 4.3389 | 3.8398 | 3.8154 | 5.5207 | 4.1628 | 4.3677 | H10 | 2.0804 | 2.6182 | 1.1106 | 4.0263 | 2.1721 | 4.9233 | 2.7892 | 2.9611 | 2.3947 | | 1.7859 | 4.1977 | 4.6552 | 3.0715 | 2.5111 | 5.9184 | 4.7384 | 5.1805 | 3.8029 | 3.1310 | 2.6150 | H11 | 2.0797 | 2.6345 | 1.1088 | 4.0232 | 2.1734 | 4.4292 | 3.5053 | 2.3947 | 3.0128 | 1.7859 | | 4.5512 | 4.7029 | 2.4998 | 2.4308 | 5.5207 | 4.1628 | 4.3677 | 4.3389 | 3.8398 | 3.8154 | H12 | 2.6281 | 2.1415 | 3.9684 | 1.1014 | 4.8708 | 2.1704 | 4.8764 | 3.0715 | 2.4998 | 4.1977 | 4.5512 | | 1.7662 | 5.0168 | 5.8734 | 2.5112 | 2.5355 | 3.0907 | 5.8091 | 4.0820 | 5.1343 | H13 | 3.3345 | 2.1341 | 4.4892 | 1.1018 | 5.7275 | 2.1722 | 6.0957 | 2.5111 | 2.4308 | 4.6552 | 4.7029 | 1.7662 | | 5.8734 | 6.5938 | 2.5269 | 3.0874 | 2.5269 | 7.1014 | 5.4924 | 6.2944 | H14 | 2.6281 | 3.9684 | 2.1415 | 4.8708 | 1.1014 | 4.8764 | 2.1704 | 4.1977 | 4.5512 | 3.0715 | 2.4998 | 5.0168 | 5.8734 | | 1.7662 | 5.8091 | 4.0820 | 5.1343 | 2.5112 | 2.5355 | 3.0907 | H15 | 3.3345 | 4.4892 | 2.1341 | 5.7275 | 1.1018 | 6.0957 | 2.1722 | 4.6552 | 4.7029 | 2.5111 | 2.4308 | 5.8734 | 6.5938 | 1.7662 | | 7.1014 | 5.4924 | 6.2944 | 2.5269 | 3.0874 | 2.5269 | H16 | 3.9582 | 3.5031 | 5.3138 | 2.1833 | 6.1042 | 1.0987 | 6.4461 | 3.8029 | 4.3389 | 5.9184 | 5.5207 | 2.5112 | 2.5269 | 5.8091 | 7.1014 | | 1.7781 | 1.7760 | 7.2186 | 5.6647 | 7.0133 | H17 | 2.6720 | 2.8088 | 3.9328 | 2.1757 | 4.5005 | 1.0977 | 4.9614 | 3.1310 | 3.8398 | 4.7384 | 4.1628 | 2.5355 | 3.0874 | 4.0820 | 5.4924 | 1.7781 | | 1.7755 | 5.6647 | 4.2883 | 5.6720 | H18 | 3.3999 | 2.8282 | 4.4935 | 2.1812 | 5.4516 | 1.1005 | 6.1935 | 2.6150 | 3.8154 | 5.1805 | 4.3677 | 3.0907 | 2.5269 | 5.1343 | 6.2944 | 1.7760 | 1.7755 | | 7.0133 | 5.6720 | 6.7687 | H19 | 3.9582 | 5.3138 | 3.5031 | 6.1042 | 2.1833 | 6.4461 | 1.0987 | 5.9184 | 5.5207 | 3.8029 | 4.3389 | 5.8091 | 7.1014 | 2.5112 | 2.5269 | 7.2186 | 5.6647 | 7.0133 | | 1.7781 | 1.7760 | H20 | 2.6720 | 3.9328 | 2.8088 | 4.5005 | 2.1757 | 4.9614 | 1.0977 | 4.7384 | 4.1628 | 3.1310 | 3.8398 | 4.0820 | 5.4924 | 2.5355 | 3.0874 | 5.6647 | 4.2883 | 5.6720 | 1.7781 | | 1.7755 | H21 | 3.3999 | 4.4935 | 2.8282 | 5.4516 | 2.1812 | 6.1935 | 1.1005 | 5.1805 | 4.3677 | 2.6150 | 3.8154 | 5.1343 | 6.2944 | 3.0907 | 2.5269 | 7.0133 | 5.6720 | 6.7687 | 1.7760 | 1.7755 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C2 |
C4 |
109.227 |
|
O1 |
C2 |
H8 |
109.841 |
| O1 |
C2 |
H9 |
109.895 |
|
O1 |
C3 |
C5 |
109.227 |
| O1 |
C3 |
H10 |
109.841 |
|
O1 |
C3 |
H11 |
109.895 |
| C2 |
O1 |
C3 |
112.602 |
|
C2 |
C4 |
C6 |
113.416 |
| C2 |
C4 |
H12 |
108.397 |
|
C2 |
C4 |
H13 |
107.814 |
| C3 |
C5 |
C7 |
113.416 |
|
C3 |
C5 |
H14 |
108.397 |
| C3 |
C5 |
H15 |
107.814 |
|
C4 |
C2 |
H8 |
110.247 |
| C4 |
C2 |
H9 |
110.453 |
|
C4 |
C6 |
H16 |
111.310 |
| C4 |
C6 |
H17 |
110.769 |
|
C4 |
C6 |
H18 |
111.039 |
| C5 |
C3 |
H10 |
110.247 |
|
C5 |
C3 |
H11 |
110.453 |
| C5 |
C7 |
H19 |
111.310 |
|
C5 |
C7 |
H20 |
110.769 |
| C5 |
C7 |
H21 |
111.039 |
|
C6 |
C4 |
H12 |
110.128 |
| C6 |
C4 |
H13 |
110.250 |
|
C7 |
C5 |
H14 |
110.128 |
| C7 |
C5 |
H15 |
110.250 |
|
H8 |
C2 |
H9 |
107.158 |
| H10 |
C3 |
H11 |
107.158 |
|
H12 |
C4 |
H13 |
106.574 |
| H14 |
C5 |
H15 |
106.574 |
|
H16 |
C6 |
H17 |
108.109 |
| H16 |
C6 |
H18 |
107.714 |
|
H17 |
C6 |
H18 |
107.751 |
| H19 |
C7 |
H20 |
108.109 |
|
H19 |
C7 |
H21 |
107.714 |
| H20 |
C7 |
H21 |
107.751 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.262 |
|
|
|
| 2 |
C |
0.036 |
|
|
|
| 3 |
C |
0.036 |
|
|
|
| 4 |
C |
-0.148 |
|
|
|
| 5 |
C |
-0.148 |
|
|
|
| 6 |
C |
-0.290 |
|
|
|
| 7 |
C |
-0.290 |
|
|
|
| 8 |
H |
0.039 |
|
|
|
| 9 |
H |
0.052 |
|
|
|
| 10 |
H |
0.039 |
|
|
|
| 11 |
H |
0.052 |
|
|
|
| 12 |
H |
0.084 |
|
|
|
| 13 |
H |
0.079 |
|
|
|
| 14 |
H |
0.084 |
|
|
|
| 15 |
H |
0.079 |
|
|
|
| 16 |
H |
0.093 |
|
|
|
| 17 |
H |
0.104 |
|
|
|
| 18 |
H |
0.083 |
|
|
|
| 19 |
H |
0.093 |
|
|
|
| 20 |
H |
0.104 |
|
|
|
| 21 |
H |
0.083 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.873 |
0.873 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-47.600 |
0.391 |
0.000 |
| y |
0.391 |
-44.529 |
0.000 |
| z |
0.000 |
0.000 |
-46.691 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.990 |
0.391 |
0.000 |
| y |
0.391 |
2.617 |
0.000 |
| z |
0.000 |
0.000 |
-0.627 |
|
| Polar |
|---|
| 3z2-r2 | -1.253 |
|---|
| x2-y2 | -3.071 |
|---|
| xy | 0.391 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.444 |
-0.661 |
0.000 |
| y |
-0.661 |
14.050 |
0.000 |
| z |
0.000 |
0.000 |
11.623 |
<r2> (average value of r
2) Å
2
| <r2> |
350.138 |
| (<r2>)1/2 |
18.712 |