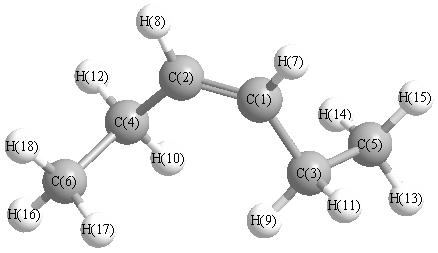
Vibrational Frequencies calculated at PBEPBE/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3058 |
3026 |
71.72 |
|
|
|
| 2 |
A |
3042 |
3010 |
0.64 |
|
|
|
| 3 |
A |
3036 |
3005 |
78.30 |
|
|
|
| 4 |
A |
3006 |
2974 |
4.14 |
|
|
|
| 5 |
A |
2966 |
2935 |
0.39 |
|
|
|
| 6 |
A |
2945 |
2915 |
6.12 |
|
|
|
| 7 |
A |
1679 |
1662 |
1.23 |
|
|
|
| 8 |
A |
1477 |
1462 |
12.38 |
|
|
|
| 9 |
A |
1467 |
1452 |
5.39 |
|
|
|
| 10 |
A |
1455 |
1440 |
0.77 |
|
|
|
| 11 |
A |
1372 |
1357 |
0.16 |
|
|
|
| 12 |
A |
1306 |
1292 |
0.13 |
|
|
|
| 13 |
A |
1275 |
1262 |
0.11 |
|
|
|
| 14 |
A |
1244 |
1231 |
0.19 |
|
|
|
| 15 |
A |
1060 |
1049 |
0.73 |
|
|
|
| 16 |
A |
1047 |
1036 |
1.53 |
|
|
|
| 17 |
A |
1000 |
989 |
0.30 |
|
|
|
| 18 |
A |
961 |
951 |
0.96 |
|
|
|
| 19 |
A |
832 |
823 |
1.82 |
|
|
|
| 20 |
A |
761 |
753 |
0.71 |
|
|
|
| 21 |
A |
503 |
498 |
0.32 |
|
|
|
| 22 |
A |
299 |
296 |
0.00 |
|
|
|
| 23 |
A |
203 |
201 |
0.01 |
|
|
|
| 24 |
A |
192 |
190 |
0.03 |
|
|
|
| 25 |
A |
60 |
60 |
0.02 |
|
|
|
| 26 |
B |
3042 |
3011 |
91.58 |
|
|
|
| 27 |
B |
3037 |
3005 |
2.08 |
|
|
|
| 28 |
B |
3031 |
2999 |
5.93 |
|
|
|
| 29 |
B |
2995 |
2964 |
7.38 |
|
|
|
| 30 |
B |
2966 |
2935 |
84.72 |
|
|
|
| 31 |
B |
2944 |
2913 |
51.21 |
|
|
|
| 32 |
B |
1476 |
1460 |
4.99 |
|
|
|
| 33 |
B |
1467 |
1452 |
11.41 |
|
|
|
| 34 |
B |
1444 |
1429 |
2.28 |
|
|
|
| 35 |
B |
1402 |
1387 |
6.14 |
|
|
|
| 36 |
B |
1370 |
1355 |
4.91 |
|
|
|
| 37 |
B |
1288 |
1275 |
8.50 |
|
|
|
| 38 |
B |
1257 |
1244 |
0.39 |
|
|
|
| 39 |
B |
1147 |
1135 |
0.23 |
|
|
|
| 40 |
B |
1065 |
1054 |
6.20 |
|
|
|
| 41 |
B |
1019 |
1009 |
3.98 |
|
|
|
| 42 |
B |
894 |
884 |
8.74 |
|
|
|
| 43 |
B |
793 |
784 |
5.80 |
|
|
|
| 44 |
B |
728 |
721 |
37.79 |
|
|
|
| 45 |
B |
552 |
546 |
6.29 |
|
|
|
| 46 |
B |
351 |
347 |
1.22 |
|
|
|
| 47 |
B |
236 |
233 |
0.06 |
|
|
|
| 48 |
B |
52 |
52 |
0.11 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 35400.1 cm
-1
Scaled (by 0.9896) Zero Point Vibrational Energy (zpe) 35032.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/6-311G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.154 |
|
|
|
| 2 |
C |
-0.154 |
|
|
|
| 3 |
C |
-0.471 |
|
|
|
| 4 |
C |
-0.471 |
|
|
|
| 5 |
C |
-0.633 |
|
|
|
| 6 |
C |
-0.633 |
|
|
|
| 7 |
H |
0.180 |
|
|
|
| 8 |
H |
0.180 |
|
|
|
| 9 |
H |
0.214 |
|
|
|
| 10 |
H |
0.214 |
|
|
|
| 11 |
H |
0.220 |
|
|
|
| 12 |
H |
0.220 |
|
|
|
| 13 |
H |
0.213 |
|
|
|
| 14 |
H |
0.220 |
|
|
|
| 15 |
H |
0.212 |
|
|
|
| 16 |
H |
0.213 |
|
|
|
| 17 |
H |
0.220 |
|
|
|
| 18 |
H |
0.212 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.230 |
0.230 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-42.275 |
0.628 |
0.000 |
| y |
0.628 |
-38.961 |
0.000 |
| z |
0.000 |
0.000 |
-39.482 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.053 |
0.628 |
0.000 |
| y |
0.628 |
1.918 |
0.000 |
| z |
0.000 |
0.000 |
1.136 |
|
| Polar |
|---|
| 3z2-r2 | 2.272 |
|---|
| x2-y2 | -3.314 |
|---|
| xy | 0.628 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.729 |
-1.180 |
0.000 |
| y |
-1.180 |
14.083 |
0.000 |
| z |
0.000 |
0.000 |
9.620 |
<r2> (average value of r
2) Å
2
| <r2> |
259.829 |
| (<r2>)1/2 |
16.119 |