Jump to
S1C2
S1C3
Vibrational Frequencies calculated at PBEPBE/3-21G*
Geometric Data calculated at PBEPBE/3-21G*
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C3
Energy calculated at PBEPBE/3-21G*
| | hartrees |
|---|
| Energy at 0K | -349.556757 |
| Energy at 298.15K | -349.557950 |
| HF Energy | -349.556757 |
| Nuclear repulsion energy | 82.837784 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3606 |
3438 |
0.12 |
|
|
|
| 2 |
A |
659 |
628 |
9.07 |
|
|
|
| 3 |
A |
212 |
202 |
37.70 |
|
|
|
| 4 |
A |
167 |
159 |
9.00 |
|
|
|
| 5 |
A |
100 |
95 |
173.68 |
|
|
|
| 6 |
B |
3607 |
3440 |
37.00 |
|
|
|
| 7 |
B |
983 |
937 |
46.83 |
|
|
|
| 8 |
B |
193 |
184 |
159.64 |
|
|
|
| 9 |
B |
148 |
141 |
113.30 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 4836.8 cm
-1
Scaled (by 0.9535) Zero Point Vibrational Energy (zpe) 4611.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/3-21G*
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Mg1 |
0.000 |
0.000 |
0.002 |
| O2 |
0.000 |
1.759 |
0.038 |
| O3 |
0.000 |
-1.759 |
0.038 |
| H4 |
-0.438 |
2.569 |
-0.315 |
| H5 |
0.438 |
-2.569 |
-0.315 |
Atom - Atom Distances (Å)
| |
Mg1 |
O2 |
O3 |
H4 |
H5 |
| Mg1 | | 1.7594 | 1.7594 | 2.6255 | 2.6255 |
O2 | 1.7594 | | 3.5181 | 0.9863 | 4.3647 | O3 | 1.7594 | 3.5181 | | 4.3647 | 0.9863 | H4 | 2.6255 | 0.9863 | 4.3647 | | 5.2125 | H5 | 2.6255 | 4.3647 | 0.9863 | 5.2125 | |
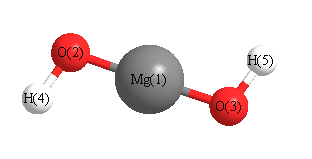 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Mg1 |
O2 |
H4 |
144.489 |
|
Mg1 |
O3 |
H5 |
144.489 |
| O2 |
Mg1 |
O3 |
177.673 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Mg |
0.559 |
|
|
|
| 2 |
O |
-0.623 |
|
|
|
| 3 |
O |
-0.623 |
|
|
|
| 4 |
H |
0.343 |
|
|
|
| 5 |
H |
0.343 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-1.124 |
1.124 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-19.959 |
-4.579 |
0.000 |
| y |
-4.579 |
-18.462 |
0.000 |
| z |
0.000 |
0.000 |
-20.444 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.506 |
-4.579 |
0.000 |
| y |
-4.579 |
1.740 |
0.000 |
| z |
0.000 |
0.000 |
-1.233 |
|
| Polar |
|---|
| 3z2-r2 | -2.467 |
|---|
| x2-y2 | -1.497 |
|---|
| xy | -4.579 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.581 |
0.116 |
0.000 |
| y |
0.116 |
5.028 |
0.000 |
| z |
0.000 |
0.000 |
3.615 |
<r2> (average value of r
2) Å
2
| <r2> |
75.570 |
| (<r2>)1/2 |
8.693 |
Jump to
S1C1
S1C2
Energy calculated at PBEPBE/3-21G*
| | hartrees |
|---|
| Energy at 0K | -349.556360 |
| Energy at 298.15K | |
| HF Energy | -349.556360 |
| Nuclear repulsion energy | 83.765537 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σg |
3675 |
3504 |
0.00 |
|
|
|
| 2 |
Σg |
675 |
644 |
0.00 |
|
|
|
| 3 |
Σu |
3677 |
3506 |
81.45 |
|
|
|
| 4 |
Σu |
1017 |
969 |
71.90 |
|
|
|
| 5 |
Πg |
143i |
136i |
0.00 |
|
|
|
| 5 |
Πg |
143i |
136i |
0.00 |
|
|
|
| 6 |
Πu |
182 |
173 |
15.79 |
|
|
|
| 6 |
Πu |
182 |
173 |
15.79 |
|
|
|
| 7 |
Πu |
136i |
129i |
248.39 |
|
|
|
| 7 |
Πu |
136i |
129i |
248.39 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 4425.1 cm
-1
Scaled (by 0.9535) Zero Point Vibrational Energy (zpe) 4219.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/3-21G*
Point Group is D∞h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Mg1 |
0.000 |
0.000 |
0.000 |
| O2 |
0.000 |
0.000 |
1.732 |
| O3 |
0.000 |
0.000 |
-1.732 |
| H4 |
0.000 |
0.000 |
2.712 |
| H5 |
0.000 |
0.000 |
-2.712 |
Atom - Atom Distances (Å)
| |
Mg1 |
O2 |
O3 |
H4 |
H5 |
| Mg1 | | 1.7321 | 1.7321 | 2.7117 | 2.7117 |
O2 | 1.7321 | | 3.4641 | 0.9796 | 4.4437 | O3 | 1.7321 | 3.4641 | | 4.4437 | 0.9796 | H4 | 2.7117 | 0.9796 | 4.4437 | | 5.4234 | H5 | 2.7117 | 4.4437 | 0.9796 | 5.4234 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Mg1 |
O2 |
H4 |
180.000 |
|
Mg1 |
O3 |
H5 |
180.000 |
| O2 |
Mg1 |
O3 |
180.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Mg |
0.593 |
|
|
|
| 2 |
O |
-0.647 |
|
|
|
| 3 |
O |
-0.647 |
|
|
|
| 4 |
H |
0.351 |
|
|
|
| 5 |
H |
0.351 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-20.897 |
0.000 |
0.000 |
| y |
0.000 |
-20.897 |
0.000 |
| z |
0.000 |
0.000 |
-14.002 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.448 |
0.000 |
0.000 |
| y |
0.000 |
-3.448 |
0.000 |
| z |
0.000 |
0.000 |
6.895 |
|
| Polar |
|---|
| 3z2-r2 | 13.791 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.465 |
0.000 |
0.000 |
| y |
0.000 |
3.465 |
0.000 |
| z |
0.000 |
0.000 |
4.857 |
<r2> (average value of r
2) Å
2
| <r2> |
74.324 |
| (<r2>)1/2 |
8.621 |