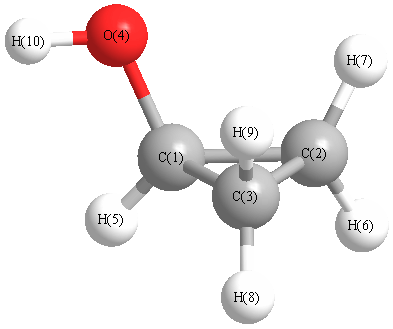
Vibrational Frequencies calculated at PBEPBE/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3330 |
3175 |
1.63 |
|
|
|
| 2 |
A |
3207 |
3058 |
8.84 |
|
|
|
| 3 |
A |
3189 |
3040 |
1.99 |
|
|
|
| 4 |
A |
3107 |
2962 |
4.05 |
|
|
|
| 5 |
A |
3096 |
2952 |
9.34 |
|
|
|
| 6 |
A |
3037 |
2896 |
44.70 |
|
|
|
| 7 |
A |
1465 |
1397 |
8.62 |
|
|
|
| 8 |
A |
1440 |
1373 |
6.90 |
|
|
|
| 9 |
A |
1399 |
1334 |
1.77 |
|
|
|
| 10 |
A |
1269 |
1210 |
52.60 |
|
|
|
| 11 |
A |
1169 |
1115 |
0.30 |
|
|
|
| 12 |
A |
1149 |
1096 |
40.48 |
|
|
|
| 13 |
A |
1144 |
1091 |
24.13 |
|
|
|
| 14 |
A |
1103 |
1052 |
1.68 |
|
|
|
| 15 |
A |
1042 |
994 |
7.74 |
|
|
|
| 16 |
A |
1014 |
967 |
38.85 |
|
|
|
| 17 |
A |
939 |
895 |
13.48 |
|
|
|
| 18 |
A |
875 |
834 |
18.61 |
|
|
|
| 19 |
A |
807 |
769 |
7.99 |
|
|
|
| 20 |
A |
782 |
745 |
3.46 |
|
|
|
| 21 |
A |
736 |
702 |
3.48 |
|
|
|
| 22 |
A |
381 |
363 |
32.99 |
|
|
|
| 23 |
A |
357 |
340 |
20.83 |
|
|
|
| 24 |
A |
331 |
316 |
95.11 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 18184.0 cm
-1
Scaled (by 0.9535) Zero Point Vibrational Energy (zpe) 17338.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.041 |
|
|
|
| 2 |
C |
-0.422 |
|
|
|
| 3 |
C |
-0.444 |
|
|
|
| 4 |
O |
-0.506 |
|
|
|
| 5 |
H |
0.196 |
|
|
|
| 6 |
H |
0.213 |
|
|
|
| 7 |
H |
0.238 |
|
|
|
| 8 |
H |
0.209 |
|
|
|
| 9 |
H |
0.223 |
|
|
|
| 10 |
H |
0.333 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.263 |
1.410 |
0.646 |
1.573 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-24.498 |
-3.381 |
0.135 |
| y |
-3.381 |
-24.188 |
-0.137 |
| z |
0.135 |
-0.137 |
-23.999 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.404 |
-3.381 |
0.135 |
| y |
-3.381 |
0.060 |
-0.137 |
| z |
0.135 |
-0.137 |
0.344 |
|
| Polar |
|---|
| 3z2-r2 | 0.688 |
|---|
| x2-y2 | -0.309 |
|---|
| xy | -3.381 |
|---|
| xz | 0.135 |
|---|
| yz | -0.137 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.166 |
-0.289 |
0.218 |
| y |
-0.289 |
4.722 |
0.031 |
| z |
0.218 |
0.031 |
4.313 |
<r2> (average value of r
2) Å
2
| <r2> |
75.017 |
| (<r2>)1/2 |
8.661 |