All results from a given calculation for C8H6 (benzocyclobutadiene)
using model chemistry: MP3/6-31+G**
19 10 17 12 22
States and conformations
| State |
Conformation |
minimum conformation |
conformer description |
state description |
| 1 |
1 |
yes |
C2V |
1A1 |
Energy calculated at MP3/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -307.423040 |
| Energy at 298.15K | |
| HF Energy | -306.345379 |
| Nuclear repulsion energy | 312.366917 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP3/6-31+G**
Geometric Data calculated at MP3/6-31+G**
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
1.451 |
-0.619 |
| C2 |
0.000 |
0.687 |
-1.846 |
| C3 |
0.000 |
-0.687 |
-1.846 |
| C4 |
0.000 |
-1.451 |
-0.619 |
| C5 |
0.000 |
-0.677 |
2.045 |
| C6 |
0.000 |
0.677 |
2.045 |
| C7 |
0.000 |
0.718 |
0.522 |
| C8 |
0.000 |
-0.718 |
0.522 |
| H9 |
0.000 |
2.531 |
-0.645 |
| H10 |
0.000 |
1.221 |
-2.786 |
| H11 |
0.000 |
-1.221 |
-2.786 |
| H12 |
0.000 |
-2.531 |
-0.645 |
| H13 |
0.000 |
-1.432 |
2.815 |
| H14 |
0.000 |
1.432 |
2.815 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
C6 |
C7 |
C8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
| C1 | | 1.4451 | 2.4643 | 2.9011 | 3.4090 | 2.7738 | 1.3558 | 2.4507 | 1.0812 | 2.1788 | 3.4396 | 3.9821 | 4.4829 | 3.4335 |
C2 | 1.4451 | | 1.3734 | 2.4643 | 4.1225 | 3.8905 | 2.3680 | 2.7533 | 2.2009 | 1.0811 | 2.1266 | 3.4347 | 5.1191 | 4.7194 | C3 | 2.4643 | 1.3734 | | 1.4451 | 3.8905 | 4.1225 | 2.7533 | 2.3680 | 3.4347 | 2.1266 | 1.0811 | 2.2009 | 4.7194 | 5.1191 | C4 | 2.9011 | 2.4643 | 1.4451 | | 2.7738 | 3.4090 | 2.4507 | 1.3558 | 3.9821 | 3.4396 | 2.1788 | 1.0812 | 3.4335 | 4.4829 | C5 | 3.4090 | 4.1225 | 3.8905 | 2.7738 | | 1.3539 | 2.0652 | 1.5232 | 4.1868 | 5.1898 | 4.8609 | 3.2673 | 1.0780 | 2.2448 | C6 | 2.7738 | 3.8905 | 4.1225 | 3.4090 | 1.3539 | | 1.5232 | 2.0652 | 3.2673 | 4.8609 | 5.1898 | 4.1868 | 2.2448 | 1.0780 | C7 | 1.3558 | 2.3680 | 2.7533 | 2.4507 | 2.0652 | 1.5232 | | 1.4366 | 2.1564 | 3.3457 | 3.8343 | 3.4530 | 3.1429 | 2.4008 | C8 | 2.4507 | 2.7533 | 2.3680 | 1.3558 | 1.5232 | 2.0652 | 1.4366 | | 3.4530 | 3.8343 | 3.3457 | 2.1564 | 2.4008 | 3.1429 | H9 | 1.0812 | 2.2009 | 3.4347 | 3.9821 | 4.1868 | 3.2673 | 2.1564 | 3.4530 | | 2.5098 | 4.3198 | 5.0628 | 5.2608 | 3.6303 | H10 | 2.1788 | 1.0811 | 2.1266 | 3.4396 | 5.1898 | 4.8609 | 3.3457 | 3.8343 | 2.5098 | | 2.4417 | 4.3198 | 6.1966 | 5.6041 | H11 | 3.4396 | 2.1266 | 1.0811 | 2.1788 | 4.8609 | 5.1898 | 3.8343 | 3.3457 | 4.3198 | 2.4417 | | 2.5098 | 5.6041 | 6.1966 | H12 | 3.9821 | 3.4347 | 2.2009 | 1.0812 | 3.2673 | 4.1868 | 3.4530 | 2.1564 | 5.0628 | 4.3198 | 2.5098 | | 3.6303 | 5.2608 | H13 | 4.4829 | 5.1191 | 4.7194 | 3.4335 | 1.0780 | 2.2448 | 3.1429 | 2.4008 | 5.2608 | 6.1966 | 5.6041 | 3.6303 | | 2.8635 | H14 | 3.4335 | 4.7194 | 5.1191 | 4.4829 | 2.2448 | 1.0780 | 2.4008 | 3.1429 | 3.6303 | 5.6041 | 6.1966 | 5.2608 | 2.8635 | |
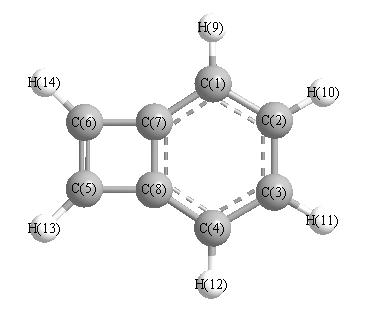 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
121.909 |
|
C1 |
C2 |
H10 |
118.481 |
| C1 |
C7 |
C6 |
148.866 |
|
C1 |
C7 |
N8 |
122.690 |
| C2 |
C1 |
C7 |
115.401 |
|
C2 |
C1 |
H9 |
120.516 |
| C2 |
C3 |
C4 |
121.909 |
|
C2 |
C3 |
H11 |
119.610 |
| C3 |
C2 |
H10 |
119.610 |
|
C3 |
C4 |
N8 |
115.401 |
| C3 |
C4 |
H12 |
120.516 |
|
C4 |
C3 |
H11 |
118.481 |
| C4 |
N8 |
C5 |
148.866 |
|
C4 |
N8 |
C7 |
122.690 |
| C5 |
C6 |
C7 |
91.556 |
|
C5 |
C6 |
H14 |
134.441 |
| C5 |
N8 |
C7 |
88.444 |
|
C6 |
C5 |
N8 |
91.556 |
| C6 |
C5 |
H13 |
134.441 |
|
C6 |
C7 |
N8 |
88.444 |
| C7 |
C1 |
H9 |
124.084 |
|
C7 |
C6 |
H14 |
134.003 |
| N8 |
C4 |
H12 |
124.084 |
|
N8 |
C5 |
H13 |
134.003 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability