All results from a given calculation for C5H8 (Ethenylcyclopropane)
using model chemistry: CCSD(T)=FULL/aug-cc-pVDZ
19 10 17 12 22
States and conformations
| State |
Conformation |
minimum conformation |
conformer description |
state description |
| 1 |
1 |
yes |
CS |
1A' |
Energy calculated at CCSD(T)=FULL/aug-cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -194.770384 |
| Energy at 298.15K | |
| HF Energy | -193.959898 |
| Nuclear repulsion energy | 161.396999 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CCSD(T)=FULL/aug-cc-pVDZ
Geometric Data calculated at CCSD(T)=FULL/aug-cc-pVDZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-0.400 |
0.292 |
0.000 |
| C2 |
0.260 |
-1.042 |
0.000 |
| C3 |
-0.399 |
-2.227 |
0.000 |
| C4 |
0.260 |
1.444 |
0.760 |
| C5 |
0.260 |
1.444 |
-0.760 |
| H6 |
-1.497 |
0.275 |
0.000 |
| H7 |
1.360 |
-1.038 |
0.000 |
| H8 |
0.141 |
-3.179 |
0.000 |
| H9 |
-1.495 |
-2.269 |
0.000 |
| H10 |
-0.396 |
2.150 |
1.278 |
| H11 |
1.203 |
1.227 |
1.271 |
| H12 |
-0.396 |
2.150 |
-1.278 |
| H13 |
1.203 |
1.227 |
-1.271 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.4883 | 2.5188 | 1.5297 | 1.5297 | 1.0964 | 2.2061 | 3.5128 | 2.7850 | 2.2554 | 2.2496 | 2.2554 | 2.2496 |
C2 | 1.4883 | | 1.3557 | 2.5996 | 2.5996 | 2.1950 | 1.1007 | 2.1400 | 2.1411 | 3.5006 | 2.7666 | 3.5006 | 2.7666 | C3 | 2.5188 | 1.3557 | | 3.8062 | 3.8062 | 2.7317 | 2.1237 | 1.0948 | 1.0968 | 4.5600 | 4.0139 | 4.5600 | 4.0139 | C4 | 1.5297 | 2.5996 | 3.8062 | | 1.5197 | 2.2424 | 2.8191 | 4.6864 | 4.1763 | 1.0940 | 1.0949 | 2.2538 | 2.2501 | C5 | 1.5297 | 2.5996 | 3.8062 | 1.5197 | | 2.2424 | 2.8191 | 4.6864 | 4.1763 | 2.2538 | 2.2501 | 1.0940 | 1.0949 | H6 | 1.0964 | 2.1950 | 2.7317 | 2.2424 | 2.2424 | | 3.1438 | 3.8224 | 2.5436 | 2.5223 | 3.1321 | 2.5223 | 3.1321 | H7 | 2.2061 | 1.1007 | 2.1237 | 2.8191 | 2.8191 | 3.1438 | | 2.4638 | 3.1096 | 3.8575 | 2.6017 | 3.8575 | 2.6017 | H8 | 3.5128 | 2.1400 | 1.0948 | 4.6864 | 4.6864 | 3.8224 | 2.4638 | | 1.8727 | 5.5066 | 4.7067 | 5.5066 | 4.7067 | H9 | 2.7850 | 2.1411 | 1.0968 | 4.1763 | 4.1763 | 2.5436 | 3.1096 | 1.8727 | | 4.7299 | 4.5952 | 4.7299 | 4.5952 | H10 | 2.2554 | 3.5006 | 4.5600 | 1.0940 | 2.2538 | 2.5223 | 3.8575 | 5.5066 | 4.7299 | | 1.8464 | 2.5551 | 3.1474 | H11 | 2.2496 | 2.7666 | 4.0139 | 1.0949 | 2.2501 | 3.1321 | 2.6017 | 4.7067 | 4.5952 | 1.8464 | | 3.1474 | 2.5428 | H12 | 2.2554 | 3.5006 | 4.5600 | 2.2538 | 1.0940 | 2.5223 | 3.8575 | 5.5066 | 4.7299 | 2.5551 | 3.1474 | | 1.8464 | H13 | 2.2496 | 2.7666 | 4.0139 | 2.2501 | 1.0949 | 3.1321 | 2.6017 | 4.7067 | 4.5952 | 3.1474 | 2.5428 | 1.8464 | |
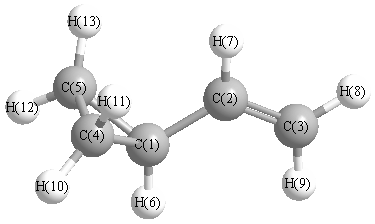 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
124.603 |
|
C1 |
C2 |
H7 |
116.085 |
| C1 |
C4 |
C5 |
60.217 |
|
C1 |
C4 |
H10 |
117.584 |
| C1 |
C4 |
H11 |
117.023 |
|
C1 |
C5 |
C4 |
60.217 |
| C1 |
C5 |
H12 |
117.584 |
|
C1 |
C5 |
H13 |
117.023 |
| C2 |
C1 |
C4 |
118.935 |
|
C2 |
C1 |
C5 |
118.935 |
| C2 |
C1 |
H6 |
115.426 |
|
C2 |
C3 |
H8 |
121.326 |
| C2 |
C3 |
H9 |
121.269 |
|
C3 |
C2 |
H7 |
119.312 |
| C4 |
C1 |
H6 |
116.298 |
|
C4 |
C5 |
H12 |
118.246 |
| C4 |
C5 |
H13 |
117.856 |
|
C5 |
C1 |
H6 |
116.298 |
| C5 |
C4 |
H10 |
118.246 |
|
C5 |
C4 |
H11 |
117.856 |
| H8 |
C3 |
H9 |
117.405 |
|
H10 |
C4 |
H11 |
115.034 |
| H12 |
C5 |
H13 |
115.034 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability