All results from a given calculation for C6H10 (1,5-Hexadiene)
using model chemistry: CCSD(T)=FULL/TZVP
19 10 17 12 22
States and conformations
| State |
Conformation |
minimum conformation |
conformer description |
state description |
| 1 |
1 |
yes |
C2 |
1Ag |
Energy calculated at CCSD(T)=FULL/TZVP
| | hartrees |
|---|
| Energy at 0K | -234.134375 |
| Energy at 298.15K | |
| HF Energy | -233.060243 |
| Nuclear repulsion energy | 212.213257 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CCSD(T)=FULL/TZVP
Geometric Data calculated at CCSD(T)=FULL/TZVP
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-0.411 |
0.651 |
-0.323 |
| C2 |
0.411 |
-0.651 |
-0.323 |
| C3 |
0.467 |
1.872 |
-0.335 |
| C4 |
-0.467 |
-1.872 |
-0.335 |
| C5 |
0.467 |
2.810 |
0.619 |
| C6 |
-0.467 |
-2.810 |
0.619 |
| H7 |
-1.070 |
0.658 |
-1.201 |
| H8 |
-1.052 |
0.669 |
0.564 |
| H9 |
1.070 |
-0.658 |
-1.201 |
| H10 |
1.052 |
-0.669 |
0.564 |
| H11 |
1.151 |
1.967 |
-1.178 |
| H12 |
-1.151 |
-1.967 |
-1.178 |
| H13 |
1.129 |
3.667 |
0.575 |
| H14 |
-0.199 |
2.744 |
1.474 |
| H15 |
-1.129 |
-3.667 |
0.575 |
| H16 |
0.199 |
-2.744 |
1.474 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
| C1 | | 1.5395 | 1.5036 | 2.5234 | 2.5138 | 3.5874 | 1.0975 | 1.0951 | 2.1624 | 2.1612 | 2.2132 | 2.8513 | 3.5030 | 2.7672 | 4.4682 | 3.8898 |
C2 | 1.5395 | | 2.5234 | 1.5036 | 3.5874 | 2.5138 | 2.1624 | 2.1612 | 1.0975 | 1.0951 | 2.8513 | 2.2132 | 4.4682 | 3.8898 | 3.5030 | 2.7672 | C3 | 1.5036 | 2.5234 | | 3.8583 | 1.3383 | 4.8683 | 2.1411 | 2.1362 | 2.7410 | 2.7581 | 1.0891 | 4.2497 | 2.1184 | 2.1162 | 5.8352 | 4.9652 | C4 | 2.5234 | 1.5036 | 3.8583 | | 4.8683 | 1.3383 | 2.7410 | 2.7581 | 2.1411 | 2.1362 | 4.2497 | 1.0891 | 5.8352 | 4.9652 | 2.1184 | 2.1162 | C5 | 2.5138 | 3.5874 | 1.3383 | 4.8683 | | 5.6968 | 3.2100 | 2.6256 | 3.9628 | 3.5283 | 2.0994 | 5.3536 | 1.0836 | 1.0856 | 6.6704 | 5.6259 | C6 | 3.5874 | 2.5138 | 4.8683 | 1.3383 | 5.6968 | | 3.9628 | 3.5283 | 3.2100 | 2.6256 | 5.3536 | 2.0994 | 6.6704 | 5.6259 | 1.0836 | 1.0856 | H7 | 1.0975 | 2.1624 | 2.1411 | 2.7410 | 3.2100 | 3.9628 | | 1.7650 | 2.5120 | 3.0626 | 2.5773 | 2.6262 | 4.1277 | 3.5024 | 4.6756 | 4.5102 | H8 | 1.0951 | 2.1612 | 2.1362 | 2.7581 | 2.6256 | 3.5283 | 1.7650 | | 3.0626 | 2.4941 | 3.0935 | 3.1608 | 3.7071 | 2.4213 | 4.3366 | 3.7477 | H9 | 2.1624 | 1.0975 | 2.7410 | 2.1411 | 3.9628 | 3.2100 | 2.5120 | 3.0626 | | 1.7650 | 2.6262 | 2.5773 | 4.6756 | 4.5102 | 4.1277 | 3.5024 | H10 | 2.1612 | 1.0951 | 2.7581 | 2.1362 | 3.5283 | 2.6256 | 3.0626 | 2.4941 | 1.7650 | | 3.1608 | 3.0935 | 4.3366 | 3.7477 | 3.7071 | 2.4213 | H11 | 2.2132 | 2.8513 | 1.0891 | 4.2497 | 2.0994 | 5.3536 | 2.5773 | 3.0935 | 2.6262 | 3.1608 | | 4.5570 | 2.4416 | 3.0755 | 6.3247 | 5.4892 | H12 | 2.8513 | 2.2132 | 4.2497 | 1.0891 | 5.3536 | 2.0994 | 2.6262 | 3.1608 | 2.5773 | 3.0935 | 4.5570 | | 6.3247 | 5.4892 | 2.4416 | 3.0755 | H13 | 3.5030 | 4.4682 | 2.1184 | 5.8352 | 1.0836 | 6.6704 | 4.1277 | 3.7071 | 4.6756 | 4.3366 | 2.4416 | 6.3247 | | 1.8501 | 7.6731 | 6.5402 | H14 | 2.7672 | 3.8898 | 2.1162 | 4.9652 | 1.0856 | 5.6259 | 3.5024 | 2.4213 | 4.5102 | 3.7477 | 3.0755 | 5.4892 | 1.8501 | | 6.5402 | 5.5028 | H15 | 4.4682 | 3.5030 | 5.8352 | 2.1184 | 6.6704 | 1.0836 | 4.6756 | 4.3366 | 4.1277 | 3.7071 | 6.3247 | 2.4416 | 7.6731 | 6.5402 | | 1.8501 | H16 | 3.8898 | 2.7672 | 4.9652 | 2.1162 | 5.6259 | 1.0856 | 4.5102 | 3.7477 | 3.5024 | 2.4213 | 5.4892 | 3.0755 | 6.5402 | 5.5028 | 1.8501 | |
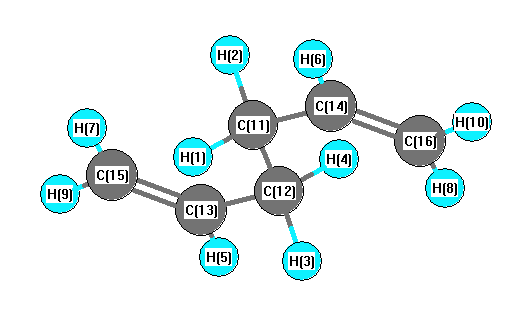 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C4 |
112.036 |
|
C1 |
C2 |
H9 |
109.018 |
| C1 |
C2 |
H10 |
109.063 |
|
C1 |
C3 |
C5 |
124.296 |
| C1 |
C3 |
H11 |
116.307 |
|
C2 |
C1 |
C3 |
112.036 |
| C2 |
C1 |
H7 |
109.018 |
|
C2 |
C1 |
H8 |
109.063 |
| C2 |
C4 |
C6 |
124.296 |
|
C2 |
C4 |
H12 |
116.307 |
| C3 |
C1 |
H7 |
109.816 |
|
C3 |
C1 |
H8 |
109.570 |
| C3 |
C5 |
H13 |
121.660 |
|
C3 |
C5 |
H14 |
121.285 |
| C4 |
C2 |
H9 |
109.816 |
|
C4 |
C2 |
H10 |
109.570 |
| C4 |
C6 |
H15 |
121.660 |
|
C4 |
C6 |
H16 |
121.285 |
| C5 |
C3 |
H11 |
119.388 |
|
C6 |
C4 |
H12 |
119.388 |
| H7 |
C1 |
H8 |
107.213 |
|
H9 |
C2 |
H10 |
107.213 |
| H13 |
C5 |
H14 |
117.055 |
|
H15 |
C6 |
H16 |
117.055 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability