All results from a given calculation for C5H6O (3-Methylfuran)
using model chemistry: CCSD(T)=FULL/TZVP
19 10 17 12 22
States and conformations
| State |
Conformation |
minimum conformation |
conformer description |
state description |
| 1 |
1 |
yes |
CS |
1A' |
Energy calculated at CCSD(T)=FULL/TZVP
| | hartrees |
|---|
| Energy at 0K | -268.829566 |
| Energy at 298.15K | |
| HF Energy | -267.748529 |
| Nuclear repulsion energy | 220.651746 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CCSD(T)=FULL/TZVP
Geometric Data calculated at CCSD(T)=FULL/TZVP
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
-0.699 |
-1.465 |
0.000 |
| C2 |
-1.080 |
-0.149 |
0.000 |
| C3 |
0.667 |
-1.465 |
0.000 |
| C4 |
0.000 |
0.680 |
0.000 |
| C5 |
1.150 |
-0.194 |
0.000 |
| C6 |
0.000 |
2.180 |
0.000 |
| H7 |
-2.143 |
0.030 |
0.000 |
| H8 |
1.134 |
-2.436 |
0.000 |
| H9 |
2.189 |
0.098 |
0.000 |
| H10 |
-1.022 |
2.567 |
0.000 |
| H11 |
0.509 |
2.573 |
0.884 |
| H12 |
0.509 |
2.573 |
-0.884 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
C4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
| O1 | | 1.3706 | 1.3663 | 2.2562 | 2.2441 | 3.7116 | 2.0786 | 2.0742 | 3.2841 | 4.0446 | 4.3073 | 4.3073 |
C2 | 1.3706 | | 2.1875 | 1.3614 | 2.2305 | 2.5669 | 1.0777 | 3.1831 | 3.2785 | 2.7157 | 3.2738 | 3.2738 | C3 | 1.3663 | 2.1875 | | 2.2461 | 1.3599 | 3.7053 | 3.1830 | 1.0770 | 2.1814 | 4.3708 | 4.1370 | 4.1370 | C4 | 2.2562 | 1.3614 | 2.2461 | | 1.4440 | 1.5000 | 2.2393 | 3.3153 | 2.2651 | 2.1455 | 2.1509 | 2.1509 | C5 | 2.2441 | 2.2305 | 1.3599 | 1.4440 | | 2.6374 | 3.3004 | 2.2419 | 1.0793 | 3.5120 | 2.9746 | 2.9746 | C6 | 3.7116 | 2.5669 | 3.7053 | 1.5000 | 2.6374 | | 3.0354 | 4.7526 | 3.0212 | 1.0925 | 1.0935 | 1.0935 | H7 | 2.0786 | 1.0777 | 3.1830 | 2.2393 | 3.3004 | 3.0354 | | 4.1010 | 4.3326 | 2.7731 | 3.7795 | 3.7795 | H8 | 2.0742 | 3.1831 | 1.0770 | 3.3153 | 2.2419 | 4.7526 | 4.1010 | | 2.7441 | 5.4467 | 5.1245 | 5.1245 | H9 | 3.2841 | 3.2785 | 2.1814 | 2.2651 | 1.0793 | 3.0212 | 4.3326 | 2.7441 | | 4.0502 | 3.1196 | 3.1196 | H10 | 4.0446 | 2.7157 | 4.3708 | 2.1455 | 3.5120 | 1.0925 | 2.7731 | 5.4467 | 4.0502 | | 1.7681 | 1.7681 | H11 | 4.3073 | 3.2738 | 4.1370 | 2.1509 | 2.9746 | 1.0935 | 3.7795 | 5.1245 | 3.1196 | 1.7681 | | 1.7681 | H12 | 4.3073 | 3.2738 | 4.1370 | 2.1509 | 2.9746 | 1.0935 | 3.7795 | 5.1245 | 3.1196 | 1.7681 | 1.7681 | |
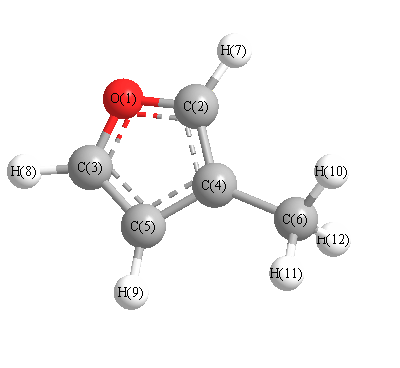 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C2 |
C4 |
111.351 |
|
O1 |
C2 |
H7 |
115.685 |
| O1 |
C3 |
C5 |
110.810 |
|
O1 |
C3 |
H8 |
115.683 |
| C2 |
O1 |
C3 |
106.124 |
|
C2 |
C4 |
C5 |
105.290 |
| C2 |
C4 |
C6 |
127.487 |
|
C3 |
C5 |
C4 |
106.426 |
| C3 |
C5 |
H9 |
126.458 |
|
C4 |
C2 |
H7 |
132.964 |
| C4 |
C5 |
H9 |
127.116 |
|
C4 |
C6 |
H10 |
110.723 |
| C4 |
C6 |
H11 |
111.092 |
|
C4 |
C6 |
H12 |
111.092 |
| C5 |
C3 |
H8 |
133.507 |
|
C5 |
C4 |
C6 |
127.223 |
| H10 |
C6 |
H11 |
107.957 |
|
H10 |
C6 |
H12 |
107.957 |
| H11 |
C6 |
H12 |
107.883 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability