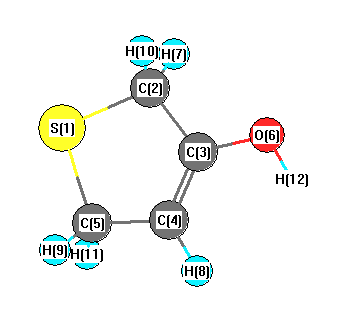
Vibrational Frequencies calculated at B3LYP/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3738 |
3589 |
28.42 |
|
|
|
| 2 |
A |
3183 |
3056 |
17.79 |
|
|
|
| 3 |
A |
3099 |
2976 |
9.94 |
|
|
|
| 4 |
A |
3078 |
2956 |
16.90 |
|
|
|
| 5 |
A |
3062 |
2941 |
34.19 |
|
|
|
| 6 |
A |
3047 |
2926 |
49.61 |
|
|
|
| 7 |
A |
1753 |
1684 |
99.66 |
|
|
|
| 8 |
A |
1529 |
1468 |
3.97 |
|
|
|
| 9 |
A |
1522 |
1461 |
3.77 |
|
|
|
| 10 |
A |
1437 |
1380 |
1.73 |
|
|
|
| 11 |
A |
1299 |
1248 |
10.48 |
|
|
|
| 12 |
A |
1290 |
1239 |
54.65 |
|
|
|
| 13 |
A |
1240 |
1191 |
2.07 |
|
|
|
| 14 |
A |
1181 |
1135 |
193.48 |
|
|
|
| 15 |
A |
1158 |
1112 |
0.01 |
|
|
|
| 16 |
A |
1140 |
1095 |
3.84 |
|
|
|
| 17 |
A |
1027 |
986 |
13.70 |
|
|
|
| 18 |
A |
982 |
943 |
0.56 |
|
|
|
| 19 |
A |
914 |
877 |
1.26 |
|
|
|
| 20 |
A |
894 |
858 |
0.06 |
|
|
|
| 21 |
A |
775 |
745 |
44.52 |
|
|
|
| 22 |
A |
773 |
743 |
11.33 |
|
|
|
| 23 |
A |
716 |
688 |
1.81 |
|
|
|
| 24 |
A |
601 |
577 |
0.28 |
|
|
|
| 25 |
A |
488 |
468 |
2.24 |
|
|
|
| 26 |
A |
478 |
459 |
0.45 |
|
|
|
| 27 |
A |
409 |
393 |
109.97 |
|
|
|
| 28 |
A |
380 |
365 |
12.06 |
|
|
|
| 29 |
A |
233 |
224 |
4.55 |
|
|
|
| 30 |
A |
87 |
84 |
3.58 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20757.2 cm
-1
Scaled (by 0.9603) Zero Point Vibrational Energy (zpe) 19933.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
S |
0.087 |
|
|
|
| 2 |
C |
-0.464 |
|
|
|
| 3 |
C |
0.346 |
|
|
|
| 4 |
C |
-0.174 |
|
|
|
| 5 |
C |
-0.450 |
|
|
|
| 6 |
O |
-0.615 |
|
|
|
| 7 |
H |
0.190 |
|
|
|
| 8 |
H |
0.124 |
|
|
|
| 9 |
H |
0.177 |
|
|
|
| 10 |
H |
0.190 |
|
|
|
| 11 |
H |
0.177 |
|
|
|
| 12 |
H |
0.411 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.972 |
1.864 |
-0.002 |
2.713 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |