All results from a given calculation for C4H8 (methylcyclopropane)
using model chemistry: CCSD=FULL/aug-cc-pVTZ
19 10 17 12 22
States and conformations
| State |
Conformation |
minimum conformation |
conformer description |
state description |
| 1 |
1 |
yes |
CS |
1A' |
Energy calculated at CCSD=FULL/aug-cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -156.970357 |
| Energy at 298.15K | |
| HF Energy | -156.152129 |
| Nuclear repulsion energy | 124.991046 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CCSD=FULL/aug-cc-pVTZ
Geometric Data calculated at CCSD=FULL/aug-cc-pVTZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.348 |
0.406 |
0.000 |
| C2 |
-0.884 |
1.263 |
0.000 |
| H3 |
1.284 |
0.943 |
0.000 |
| C4 |
0.348 |
-0.889 |
0.752 |
| C5 |
0.348 |
-0.889 |
-0.752 |
| H6 |
1.251 |
-1.192 |
1.258 |
| H7 |
1.251 |
-1.192 |
-1.258 |
| H8 |
-0.565 |
-1.170 |
1.252 |
| H9 |
-0.565 |
-1.170 |
-1.252 |
| H10 |
-1.777 |
0.637 |
0.000 |
| H11 |
-0.924 |
1.900 |
0.883 |
| H12 |
-0.924 |
1.900 |
-0.883 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
H3 |
C4 |
C5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
| C1 | | 1.5010 | 1.0791 | 1.4975 | 1.4975 | 2.2245 | 2.2245 | 2.2102 | 2.2102 | 2.1380 | 2.1521 | 2.1521 |
C2 | 1.5010 | | 2.1914 | 2.5912 | 2.5912 | 3.4876 | 3.4876 | 2.7544 | 2.7544 | 1.0905 | 1.0898 | 1.0898 | H3 | 1.0791 | 2.1914 | | 2.1907 | 2.1907 | 2.4784 | 2.4784 | 3.0747 | 3.0747 | 3.0764 | 2.5632 | 2.5632 | C4 | 1.4975 | 2.5912 | 2.1907 | | 1.5050 | 1.0775 | 2.2241 | 1.0785 | 2.2205 | 2.7226 | 3.0683 | 3.4745 | C5 | 1.4975 | 2.5912 | 2.1907 | 1.5050 | | 2.2241 | 1.0775 | 2.2205 | 1.0785 | 2.7226 | 3.4745 | 3.0683 | H6 | 2.2245 | 3.4876 | 2.4784 | 1.0775 | 2.2241 | | 2.5153 | 1.8162 | 3.0976 | 3.7542 | 3.7986 | 4.3442 | H7 | 2.2245 | 3.4876 | 2.4784 | 2.2241 | 1.0775 | 2.5153 | | 3.0976 | 1.8162 | 3.7542 | 4.3442 | 3.7986 | H8 | 2.2102 | 2.7544 | 3.0747 | 1.0785 | 2.2205 | 1.8162 | 3.0976 | | 2.5033 | 2.5101 | 3.1130 | 3.7566 | H9 | 2.2102 | 2.7544 | 3.0747 | 2.2205 | 1.0785 | 3.0976 | 1.8162 | 2.5033 | | 2.5101 | 3.7566 | 3.1130 | H10 | 2.1380 | 1.0905 | 3.0764 | 2.7226 | 2.7226 | 3.7542 | 3.7542 | 2.5101 | 2.5101 | | 1.7616 | 1.7616 | H11 | 2.1521 | 1.0898 | 2.5632 | 3.0683 | 3.4745 | 3.7986 | 4.3442 | 3.1130 | 3.7566 | 1.7616 | | 1.7662 | H12 | 2.1521 | 1.0898 | 2.5632 | 3.4745 | 3.0683 | 4.3442 | 3.7986 | 3.7566 | 3.1130 | 1.7616 | 1.7662 | |
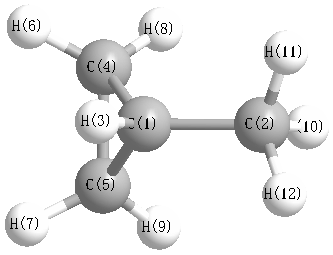 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
H10 |
110.175 |
|
C1 |
C2 |
H11 |
111.345 |
| C1 |
C2 |
H12 |
111.345 |
|
C1 |
C4 |
H5 |
59.834 |
| C1 |
C4 |
H6 |
118.598 |
|
C1 |
C4 |
H8 |
117.259 |
| C1 |
H5 |
C4 |
59.834 |
|
C1 |
H5 |
H7 |
118.598 |
| C1 |
H5 |
H9 |
117.259 |
|
C2 |
C1 |
C3 |
115.304 |
| C2 |
C1 |
C4 |
119.577 |
|
C2 |
C1 |
H5 |
119.577 |
| C3 |
C1 |
C4 |
115.515 |
|
C3 |
C1 |
H5 |
115.515 |
| C4 |
C1 |
H5 |
60.332 |
|
C4 |
H5 |
H7 |
117.957 |
| C4 |
H5 |
H9 |
117.569 |
|
H5 |
C4 |
H6 |
117.957 |
| H5 |
C4 |
H8 |
117.569 |
|
H6 |
C4 |
H8 |
114.781 |
| H7 |
H5 |
H9 |
114.781 |
|
H10 |
C2 |
H11 |
107.794 |
| H10 |
C2 |
H12 |
107.794 |
|
H11 |
C2 |
H12 |
108.247 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability