Jump to
S1C2
S1C3
Energy calculated at QCISD(TQ)/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -100.084973 |
| Energy at 298.15K | -100.083902 |
| HF Energy | -99.778198 |
| Nuclear repulsion energy | 27.111253 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at QCISD(TQ)/cc-pVDZ
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
0.000 |
0.000 |
-2.112 |
| C2 |
0.000 |
0.000 |
-0.152 |
| N3 |
0.000 |
0.000 |
1.036 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 1.9603 | 3.1478 |
C2 | 1.9603 | | 1.1875 | N3 | 3.1478 | 1.1875 | |
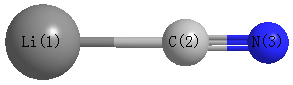 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
180.000 |
|
Li1 |
N3 |
C2 |
0.000 |
| C2 |
Li1 |
N3 |
0.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C3
Energy calculated at QCISD(TQ)/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -100.091264 |
| Energy at 298.15K | -100.090468 |
| HF Energy | -99.787764 |
| Nuclear repulsion energy | 28.671061 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at QCISD(TQ)/cc-pVDZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
1.478 |
-0.600 |
0.000 |
| C2 |
-0.739 |
-0.371 |
0.000 |
| N3 |
0.000 |
0.575 |
0.000 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 2.2292 | 1.8881 |
C2 | 2.2292 | | 1.2004 | N3 | 1.8881 | 1.2004 | |
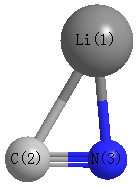 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
57.882 |
|
Li1 |
N3 |
C2 |
89.536 |
| C2 |
Li1 |
N3 |
32.581 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
Energy calculated at QCISD(TQ)/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -100.089214 |
| Energy at 298.15K | |
| HF Energy | -99.790648 |
| Nuclear repulsion energy | 27.836520 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at QCISD(TQ)/cc-pVDZ
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
0.000 |
0.000 |
1.924 |
| C2 |
0.000 |
0.000 |
-1.089 |
| N3 |
0.000 |
0.000 |
0.109 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 3.0128 | 1.8151 |
C2 | 3.0128 | | 1.1977 | N3 | 1.8151 | 1.1977 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
0.000 |
|
Li1 |
N3 |
C2 |
180.000 |
| C2 |
Li1 |
N3 |
0.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability