Vibrational Frequencies calculated at PBEPBEultrafine/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3688 |
3646 |
19.10 |
|
|
|
| 2 |
A |
3160 |
3125 |
10.60 |
|
|
|
| 3 |
A |
3144 |
3109 |
2.03 |
|
|
|
| 4 |
A |
3071 |
3037 |
6.50 |
|
|
|
| 5 |
A |
3062 |
3028 |
13.49 |
|
|
|
| 6 |
A |
3029 |
2995 |
31.66 |
|
|
|
| 7 |
A |
1445 |
1429 |
16.36 |
|
|
|
| 8 |
A |
1399 |
1384 |
2.73 |
|
|
|
| 9 |
A |
1358 |
1343 |
7.47 |
|
|
|
| 10 |
A |
1252 |
1238 |
49.76 |
|
|
|
| 11 |
A |
1192 |
1179 |
61.44 |
|
|
|
| 12 |
A |
1151 |
1138 |
0.24 |
|
|
|
| 13 |
A |
1146 |
1133 |
15.04 |
|
|
|
| 14 |
A |
1080 |
1068 |
1.70 |
|
|
|
| 15 |
A |
1018 |
1007 |
2.19 |
|
|
|
| 16 |
A |
996 |
985 |
28.45 |
|
|
|
| 17 |
A |
956 |
946 |
12.56 |
|
|
|
| 18 |
A |
903 |
893 |
18.67 |
|
|
|
| 19 |
A |
809 |
800 |
10.89 |
|
|
|
| 20 |
A |
791 |
782 |
6.51 |
|
|
|
| 21 |
A |
729 |
721 |
3.51 |
|
|
|
| 22 |
A |
395 |
391 |
8.01 |
|
|
|
| 23 |
A |
391 |
386 |
10.73 |
|
|
|
| 24 |
A |
291 |
287 |
99.46 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 18229.1 cm
-1
Scaled (by 0.9888) Zero Point Vibrational Energy (zpe) 18024.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
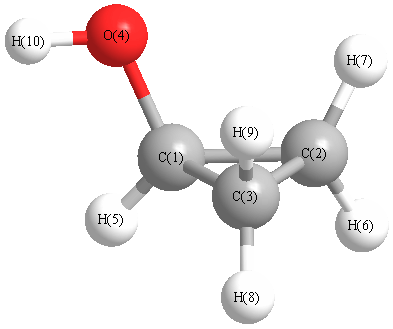
Charges from optimized geometry at PBEPBEultrafine/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.295 |
|
|
|
| 2 |
C |
-0.552 |
|
|
|
| 3 |
C |
-0.586 |
|
|
|
| 4 |
O |
-0.506 |
|
|
|
| 5 |
H |
0.300 |
|
|
|
| 6 |
H |
0.243 |
|
|
|
| 7 |
H |
0.214 |
|
|
|
| 8 |
H |
0.217 |
|
|
|
| 9 |
H |
0.235 |
|
|
|
| 10 |
H |
0.141 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.446 |
1.234 |
0.668 |
1.472 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-25.464 |
-3.129 |
-0.012 |
| y |
-3.129 |
-24.798 |
-0.133 |
| z |
-0.012 |
-0.133 |
-24.696 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.717 |
-3.129 |
-0.012 |
| y |
-3.129 |
0.283 |
-0.133 |
| z |
-0.012 |
-0.133 |
0.435 |
|
| Polar |
|---|
| 3z2-r2 | 0.870 |
|---|
| x2-y2 | -0.667 |
|---|
| xy | -3.129 |
|---|
| xz | -0.012 |
|---|
| yz | -0.133 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.987 |
-0.118 |
-0.056 |
| y |
-0.118 |
6.506 |
-0.016 |
| z |
-0.056 |
-0.016 |
5.949 |
<r2> (average value of r
2) Å
2
| <r2> |
74.203 |
| (<r2>)1/2 |
8.614 |