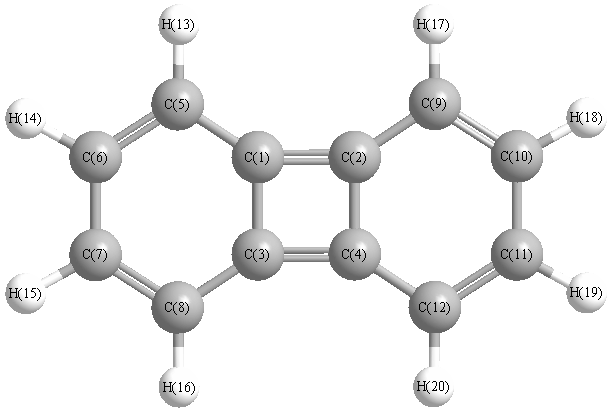
Vibrational Frequencies calculated at PBEPBEultrafine/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3129 |
3093 |
0.00 |
|
|
|
| 2 |
Ag |
3114 |
3078 |
0.00 |
|
|
|
| 3 |
Ag |
1668 |
1649 |
0.00 |
|
|
|
| 4 |
Ag |
1478 |
1461 |
0.00 |
|
|
|
| 5 |
Ag |
1391 |
1376 |
0.00 |
|
|
|
| 6 |
Ag |
1164 |
1151 |
0.00 |
|
|
|
| 7 |
Ag |
1109 |
1097 |
0.00 |
|
|
|
| 8 |
Ag |
994 |
983 |
0.00 |
|
|
|
| 9 |
Ag |
762 |
754 |
0.00 |
|
|
|
| 10 |
Ag |
391 |
386 |
0.00 |
|
|
|
| 11 |
Au |
903 |
893 |
0.00 |
|
|
|
| 12 |
Au |
848 |
838 |
0.00 |
|
|
|
| 13 |
Au |
706 |
698 |
0.00 |
|
|
|
| 14 |
Au |
542 |
536 |
0.00 |
|
|
|
| 15 |
Au |
139 |
137 |
0.00 |
|
|
|
| 16 |
B1g |
903 |
893 |
0.00 |
|
|
|
| 17 |
B1g |
837 |
828 |
0.00 |
|
|
|
| 18 |
B1g |
628 |
621 |
0.00 |
|
|
|
| 19 |
B1g |
429 |
425 |
0.00 |
|
|
|
| 20 |
B1u |
3128 |
3092 |
31.92 |
|
|
|
| 21 |
B1u |
3113 |
3078 |
10.77 |
|
|
|
| 22 |
B1u |
1585 |
1567 |
0.92 |
|
|
|
| 23 |
B1u |
1415 |
1399 |
29.67 |
|
|
|
| 24 |
B1u |
1304 |
1290 |
22.09 |
|
|
|
| 25 |
B1u |
1156 |
1143 |
5.35 |
|
|
|
| 26 |
B1u |
1019 |
1007 |
0.19 |
|
|
|
| 27 |
B1u |
977 |
966 |
20.81 |
|
|
|
| 28 |
B1u |
608 |
601 |
3.84 |
|
|
|
| 29 |
B2g |
872 |
862 |
0.00 |
|
|
|
| 30 |
B2g |
722 |
714 |
0.00 |
|
|
|
| 31 |
B2g |
409 |
404 |
0.00 |
|
|
|
| 32 |
B2g |
305 |
301 |
0.00 |
|
|
|
| 33 |
B2u |
3122 |
3087 |
40.68 |
|
|
|
| 34 |
B2u |
3102 |
3067 |
0.06 |
|
|
|
| 35 |
B2u |
1577 |
1559 |
1.87 |
|
|
|
| 36 |
B2u |
1435 |
1419 |
4.85 |
|
|
|
| 37 |
B2u |
1254 |
1240 |
5.06 |
|
|
|
| 38 |
B2u |
1120 |
1107 |
13.77 |
|
|
|
| 39 |
B2u |
1046 |
1034 |
0.46 |
|
|
|
| 40 |
B2u |
716 |
707 |
0.38 |
|
|
|
| 41 |
B2u |
209 |
206 |
0.81 |
|
|
|
| 42 |
B3g |
3122 |
3086 |
0.00 |
|
|
|
| 43 |
B3g |
3102 |
3067 |
0.00 |
|
|
|
| 44 |
B3g |
1595 |
1577 |
0.00 |
|
|
|
| 45 |
B3g |
1437 |
1420 |
0.00 |
|
|
|
| 46 |
B3g |
1269 |
1254 |
0.00 |
|
|
|
| 47 |
B3g |
1086 |
1074 |
0.00 |
|
|
|
| 48 |
B3g |
974 |
963 |
0.00 |
|
|
|
| 49 |
B3g |
596 |
589 |
0.00 |
|
|
|
| 50 |
B3g |
550 |
544 |
0.00 |
|
|
|
| 51 |
B3u |
874 |
864 |
7.45 |
|
|
|
| 52 |
B3u |
713 |
705 |
140.54 |
|
|
|
| 53 |
B3u |
354 |
350 |
2.06 |
|
|
|
| 54 |
B3u |
94 |
93 |
2.85 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 33545.3 cm
-1
Scaled (by 0.9886) Zero Point Vibrational Energy (zpe) 33162.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBEultrafine/TZVP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.019 |
|
|
|
| 2 |
C |
0.019 |
|
|
|
| 3 |
C |
0.019 |
|
|
|
| 4 |
C |
0.019 |
|
|
|
| 5 |
C |
-0.129 |
|
|
|
| 6 |
C |
-0.093 |
|
|
|
| 7 |
C |
-0.093 |
|
|
|
| 8 |
C |
-0.129 |
|
|
|
| 9 |
C |
-0.129 |
|
|
|
| 10 |
C |
-0.093 |
|
|
|
| 11 |
C |
-0.093 |
|
|
|
| 12 |
C |
-0.129 |
|
|
|
| 13 |
H |
0.103 |
|
|
|
| 14 |
H |
0.101 |
|
|
|
| 15 |
H |
0.101 |
|
|
|
| 16 |
H |
0.103 |
|
|
|
| 17 |
H |
0.103 |
|
|
|
| 18 |
H |
0.101 |
|
|
|
| 19 |
H |
0.101 |
|
|
|
| 20 |
H |
0.103 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-75.183 |
0.000 |
0.000 |
| y |
0.000 |
-60.893 |
0.000 |
| z |
0.000 |
0.000 |
-60.465 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-14.504 |
0.000 |
0.000 |
| y |
0.000 |
6.931 |
0.000 |
| z |
0.000 |
0.000 |
7.573 |
|
| Polar |
|---|
| 3z2-r2 | 15.147 |
|---|
| x2-y2 | -14.290 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.755 |
0.000 |
0.000 |
| y |
0.000 |
20.519 |
0.000 |
| z |
0.000 |
0.000 |
34.065 |
<r2> (average value of r
2) Å
2
| <r2> |
564.713 |
| (<r2>)1/2 |
23.764 |