Jump to
S1C2
S1C3
Energy calculated at PBEPBEultrafine/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -581.028241 |
| Energy at 298.15K | |
| HF Energy | -581.028241 |
| Nuclear repulsion energy | 78.495519 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBEultrafine/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2193 |
2193 |
0.00 |
424.63 |
0.11 |
0.20 |
| 2 |
Ag |
906 |
906 |
0.00 |
10.88 |
0.41 |
0.58 |
| 3 |
Ag |
574 |
574 |
0.00 |
82.62 |
0.22 |
0.35 |
| 4 |
Au |
530 |
530 |
0.00 |
0.00 |
0.00 |
0.00 |
| 5 |
B1u |
2188 |
2188 |
49.78 |
0.00 |
0.00 |
0.00 |
| 6 |
B1u |
828 |
828 |
100.96 |
0.00 |
0.00 |
0.00 |
| 7 |
B2g |
233i |
233i |
0.00 |
18.34 |
0.75 |
0.86 |
| 8 |
B2u |
2226 |
2226 |
81.04 |
0.00 |
0.00 |
0.00 |
| 9 |
B2u |
335 |
335 |
14.69 |
0.00 |
0.00 |
0.00 |
| 10 |
B3g |
2216 |
2216 |
0.00 |
214.26 |
0.75 |
0.86 |
| 11 |
B3g |
575 |
575 |
0.00 |
5.37 |
0.75 |
0.86 |
| 12 |
B3u |
488 |
488 |
0.52 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 6413.0 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 6413.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBEultrafine/Def2TZVPP
Point Group is D2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
0.000 |
1.073 |
| Si2 |
0.000 |
0.000 |
-1.073 |
| H3 |
0.000 |
1.259 |
1.863 |
| H4 |
0.000 |
-1.259 |
1.863 |
| H5 |
0.000 |
1.259 |
-1.863 |
| H6 |
0.000 |
-1.259 |
-1.863 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
H5 |
H6 |
| Si1 | | 2.1450 | 1.4871 | 1.4871 | 3.1945 | 3.1945 |
Si2 | 2.1450 | | 3.1945 | 3.1945 | 1.4871 | 1.4871 | H3 | 1.4871 | 3.1945 | | 2.5190 | 3.7265 | 4.4980 | H4 | 1.4871 | 3.1945 | 2.5190 | | 4.4980 | 3.7265 | H5 | 3.1945 | 1.4871 | 3.7265 | 4.4980 | | 2.5190 | H6 | 3.1945 | 1.4871 | 4.4980 | 3.7265 | 2.5190 | |
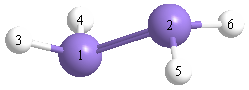 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H5 |
122.122 |
|
Si1 |
Si2 |
H6 |
122.122 |
| Si2 |
Si1 |
H3 |
122.122 |
|
Si2 |
Si1 |
H4 |
122.122 |
| H3 |
Si1 |
H4 |
115.756 |
|
H5 |
Si2 |
H6 |
115.756 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBEultrafine/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.038 |
|
|
|
| 2 |
Si |
0.038 |
|
|
|
| 3 |
H |
-0.019 |
|
|
|
| 4 |
H |
-0.019 |
|
|
|
| 5 |
H |
-0.019 |
|
|
|
| 6 |
H |
-0.019 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.650 |
0.000 |
0.000 |
| y |
0.000 |
-29.020 |
0.000 |
| z |
0.000 |
0.000 |
-27.247 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.517 |
0.000 |
0.000 |
| y |
0.000 |
0.429 |
0.000 |
| z |
0.000 |
0.000 |
3.088 |
|
| Polar |
|---|
| 3z2-r2 | 6.176 |
|---|
| x2-y2 | -2.630 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.843 |
0.000 |
0.000 |
| y |
0.000 |
8.126 |
0.000 |
| z |
0.000 |
0.000 |
14.778 |
<r2> (average value of r
2) Å
2
| <r2> |
70.745 |
| (<r2>)1/2 |
8.411 |
Jump to
S1C1
S1C3
Energy calculated at PBEPBEultrafine/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -581.029368 |
| Energy at 298.15K | |
| HF Energy | -581.029368 |
| Nuclear repulsion energy | 77.842296 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBEultrafine/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2162 |
2162 |
0.00 |
441.78 |
0.12 |
0.22 |
| 2 |
Ag |
909 |
909 |
0.00 |
19.80 |
0.49 |
0.66 |
| 3 |
Ag |
549 |
549 |
0.00 |
63.35 |
0.26 |
0.42 |
| 4 |
Ag |
311 |
311 |
0.00 |
19.74 |
0.10 |
0.18 |
| 5 |
Au |
2194 |
2194 |
108.04 |
0.00 |
0.00 |
0.00 |
| 6 |
Au |
507 |
507 |
0.00 |
0.00 |
0.00 |
0.00 |
| 7 |
Au |
325 |
325 |
12.91 |
0.00 |
0.00 |
0.00 |
| 8 |
Bg |
2183 |
2183 |
0.00 |
247.85 |
0.75 |
0.86 |
| 9 |
Bg |
587 |
587 |
0.00 |
8.51 |
0.75 |
0.86 |
| 10 |
Bu |
2159 |
2159 |
88.03 |
0.00 |
0.00 |
0.00 |
| 11 |
Bu |
876 |
876 |
152.54 |
0.00 |
0.00 |
0.00 |
| 12 |
Bu |
410 |
410 |
20.44 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 6585.3 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 6585.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBEultrafine/Def2TZVPP
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.087 |
0.000 |
| Si2 |
0.000 |
-1.087 |
0.000 |
| H3 |
0.425 |
1.803 |
1.239 |
| H4 |
0.425 |
1.803 |
-1.239 |
| H5 |
-0.425 |
-1.803 |
1.239 |
| H6 |
-0.425 |
-1.803 |
-1.239 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
H5 |
H6 |
| Si1 | | 2.1740 | 1.4932 | 1.4932 | 3.1732 | 3.1732 |
Si2 | 2.1740 | | 3.1732 | 3.1732 | 1.4932 | 1.4932 | H3 | 1.4932 | 3.1732 | | 2.4790 | 3.7050 | 4.4578 | H4 | 1.4932 | 3.1732 | 2.4790 | | 4.4578 | 3.7050 | H5 | 3.1732 | 1.4932 | 3.7050 | 4.4578 | | 2.4790 | H6 | 3.1732 | 1.4932 | 4.4578 | 3.7050 | 2.4790 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H5 |
118.653 |
|
Si1 |
Si2 |
H6 |
118.653 |
| Si2 |
Si1 |
H3 |
118.653 |
|
Si2 |
Si1 |
H4 |
118.653 |
| H3 |
Si1 |
H4 |
112.211 |
|
H5 |
Si2 |
H6 |
112.211 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBEultrafine/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.050 |
|
|
|
| 2 |
Si |
0.050 |
|
|
|
| 3 |
H |
-0.025 |
|
|
|
| 4 |
H |
-0.025 |
|
|
|
| 5 |
H |
-0.025 |
|
|
|
| 6 |
H |
-0.025 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.369 |
-0.291 |
0.000 |
| y |
-0.291 |
-27.543 |
0.000 |
| z |
0.000 |
0.000 |
-29.356 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.920 |
-0.291 |
0.000 |
| y |
-0.291 |
2.820 |
0.000 |
| z |
0.000 |
0.000 |
0.100 |
|
| Polar |
|---|
| 3z2-r2 | 0.200 |
|---|
| x2-y2 | -3.826 |
|---|
| xy | -0.291 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.020 |
-0.266 |
0.000 |
| y |
-0.266 |
15.079 |
0.000 |
| z |
0.000 |
0.000 |
8.489 |
<r2> (average value of r
2) Å
2
| <r2> |
71.334 |
| (<r2>)1/2 |
8.446 |
Jump to
S1C1
S1C2
Energy calculated at PBEPBEultrafine/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -580.995059 |
| Energy at 298.15K | |
| HF Energy | -580.995059 |
| Nuclear repulsion energy | 72.787588 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBEultrafine/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
2024 |
2024 |
286.69 |
257.41 |
0.30 |
0.47 |
| 2 |
A1 |
1582 |
1582 |
0.09 |
63.34 |
0.09 |
0.16 |
| 3 |
A1 |
822 |
822 |
16.19 |
46.59 |
0.31 |
0.47 |
| 4 |
A1 |
375 |
375 |
1.02 |
47.98 |
0.29 |
0.44 |
| 5 |
A1 |
350 |
350 |
0.01 |
8.52 |
0.72 |
0.84 |
| 6 |
A2 |
1394 |
1394 |
0.00 |
0.49 |
0.75 |
0.86 |
| 7 |
A2 |
606 |
606 |
0.00 |
0.13 |
0.75 |
0.86 |
| 8 |
B1 |
1275 |
1275 |
15.73 |
0.33 |
0.75 |
0.86 |
| 9 |
B1 |
759 |
759 |
9.84 |
22.73 |
0.75 |
0.86 |
| 10 |
B2 |
1998 |
1998 |
13.12 |
26.42 |
0.75 |
0.86 |
| 11 |
B2 |
1324 |
1324 |
751.22 |
20.54 |
0.75 |
0.86 |
| 12 |
B2 |
676 |
676 |
61.77 |
6.38 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 6591.9 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 6591.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBEultrafine/Def2TZVPP
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.321 |
-0.090 |
| Si2 |
0.000 |
-1.321 |
-0.090 |
| H3 |
-1.038 |
0.000 |
-0.183 |
| H4 |
1.038 |
0.000 |
-0.183 |
| H5 |
0.000 |
1.315 |
1.438 |
| H6 |
0.000 |
-1.315 |
1.438 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
H5 |
H6 |
| Si1 | | 2.6424 | 1.6826 | 1.6826 | 1.5281 | 3.0469 |
Si2 | 2.6424 | | 1.6826 | 1.6826 | 3.0469 | 1.5281 | H3 | 1.6826 | 1.6826 | | 2.0753 | 2.3313 | 2.3313 | H4 | 1.6826 | 1.6826 | 2.0753 | | 2.3313 | 2.3313 | H5 | 1.5281 | 3.0469 | 2.3313 | 2.3313 | | 2.6298 | H6 | 3.0469 | 1.5281 | 2.3313 | 2.3313 | 2.6298 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H5 |
30.100 |
|
Si1 |
Si2 |
H6 |
89.763 |
| Si2 |
Si1 |
H3 |
38.258 |
|
Si2 |
Si1 |
H4 |
38.258 |
| H3 |
Si1 |
H4 |
76.153 |
|
H5 |
Si2 |
H6 |
59.663 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBEultrafine/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.042 |
|
|
|
| 2 |
Si |
0.042 |
|
|
|
| 3 |
H |
-0.011 |
|
|
|
| 4 |
H |
-0.011 |
|
|
|
| 5 |
H |
-0.031 |
|
|
|
| 6 |
H |
-0.031 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.380 |
0.380 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-27.258 |
0.000 |
0.000 |
| y |
0.000 |
-31.901 |
0.000 |
| z |
0.000 |
0.000 |
-33.136 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.260 |
0.000 |
0.000 |
| y |
0.000 |
-1.704 |
0.000 |
| z |
0.000 |
0.000 |
-3.557 |
|
| Polar |
|---|
| 3z2-r2 | -7.114 |
|---|
| x2-y2 | 4.643 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.399 |
0.000 |
0.000 |
| y |
0.000 |
14.124 |
0.000 |
| z |
0.000 |
0.000 |
9.131 |
<r2> (average value of r
2) Å
2
| <r2> |
78.133 |
| (<r2>)1/2 |
8.839 |