Vibrational Frequencies calculated at PBEPBEultrafine/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3139 |
3121 |
14.32 |
|
|
|
| 2 |
A' |
3133 |
3115 |
11.29 |
|
|
|
| 3 |
A' |
3126 |
3108 |
3.11 |
|
|
|
| 4 |
A' |
3121 |
3103 |
2.71 |
|
|
|
| 5 |
A' |
3111 |
3093 |
1.07 |
|
|
|
| 6 |
A' |
1611 |
1601 |
1.87 |
|
|
|
| 7 |
A' |
1602 |
1593 |
1.60 |
|
|
|
| 8 |
A' |
1534 |
1525 |
160.97 |
|
|
|
| 9 |
A' |
1445 |
1437 |
7.38 |
|
|
|
| 10 |
A' |
1431 |
1423 |
36.21 |
|
|
|
| 11 |
A' |
1390 |
1382 |
12.19 |
|
|
|
| 12 |
A' |
1272 |
1265 |
6.00 |
|
|
|
| 13 |
A' |
1149 |
1143 |
25.01 |
|
|
|
| 14 |
A' |
1137 |
1130 |
0.55 |
|
|
|
| 15 |
A' |
1083 |
1077 |
123.38 |
|
|
|
| 16 |
A' |
1053 |
1047 |
10.31 |
|
|
|
| 17 |
A' |
1008 |
1002 |
7.38 |
|
|
|
| 18 |
A' |
986 |
980 |
0.45 |
|
|
|
| 19 |
A' |
807 |
802 |
37.68 |
|
|
|
| 20 |
A' |
661 |
658 |
5.31 |
|
|
|
| 21 |
A' |
598 |
595 |
0.08 |
|
|
|
| 22 |
A' |
428 |
426 |
1.22 |
|
|
|
| 23 |
A' |
245 |
243 |
2.23 |
|
|
|
| 24 |
A" |
991 |
985 |
0.16 |
|
|
|
| 25 |
A" |
974 |
968 |
0.01 |
|
|
|
| 26 |
A" |
939 |
933 |
2.28 |
|
|
|
| 27 |
A" |
845 |
840 |
0.00 |
|
|
|
| 28 |
A" |
759 |
755 |
34.03 |
|
|
|
| 29 |
A" |
680 |
676 |
21.37 |
|
|
|
| 30 |
A" |
458 |
455 |
1.52 |
|
|
|
| 31 |
A" |
403 |
401 |
0.00 |
|
|
|
| 32 |
A" |
233 |
232 |
0.16 |
|
|
|
| 33 |
A" |
117 |
117 |
0.07 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20734.5 cm
-1
Scaled (by 0.9942) Zero Point Vibrational Energy (zpe) 20614.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
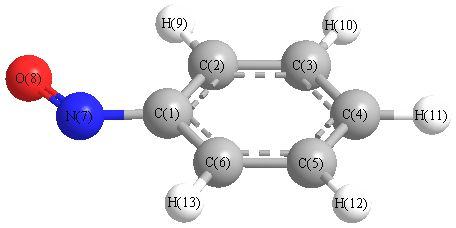
Charges from optimized geometry at PBEPBEultrafine/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.024 |
|
|
|
| 2 |
C |
0.081 |
|
|
|
| 3 |
C |
0.035 |
|
|
|
| 4 |
C |
0.036 |
|
|
|
| 5 |
C |
0.035 |
|
|
|
| 6 |
C |
0.076 |
|
|
|
| 7 |
N |
-0.036 |
|
|
|
| 8 |
O |
-0.159 |
|
|
|
| 9 |
H |
-0.014 |
|
|
|
| 10 |
H |
-0.005 |
|
|
|
| 11 |
H |
-0.005 |
|
|
|
| 12 |
H |
-0.006 |
|
|
|
| 13 |
H |
-0.015 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.098 |
-3.329 |
0.000 |
3.505 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-41.919 |
2.092 |
0.000 |
| y |
2.092 |
-47.715 |
0.000 |
| z |
0.000 |
0.000 |
-47.092 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.484 |
2.092 |
0.000 |
| y |
2.092 |
-3.209 |
0.000 |
| z |
0.000 |
0.000 |
-2.275 |
|
| Polar |
|---|
| 3z2-r2 | -4.549 |
|---|
| x2-y2 | 5.796 |
|---|
| xy | 2.092 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
13.053 |
-1.735 |
0.000 |
| y |
-1.735 |
15.721 |
0.000 |
| z |
0.000 |
0.000 |
4.218 |
<r2> (average value of r
2) Å
2
| <r2> |
252.487 |
| (<r2>)1/2 |
15.890 |