Jump to
S1C2
Energy calculated at G2
| | hartrees |
|---|
| Energy at 0K | -187.721335 |
| Energy at 298.15K | -187.715200 |
| HF Energy | -186.871072 |
| Nuclear repulsion energy | 102.629084 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3869 |
3645 |
25.85 |
|
|
|
| 2 |
A |
3773 |
3555 |
5.86 |
|
|
|
| 3 |
A |
2558 |
2410 |
0.04 |
|
|
|
| 4 |
A |
1825 |
1720 |
14.53 |
|
|
|
| 5 |
A |
1327 |
1251 |
0.02 |
|
|
|
| 6 |
A |
876 |
825 |
9.21 |
|
|
|
| 7 |
A |
684 |
644 |
166.53 |
|
|
|
| 8 |
A |
505 |
476 |
8.56 |
|
|
|
| 9 |
A |
391 |
368 |
32.27 |
|
|
|
| 10 |
A |
251 |
236 |
20.14 |
|
|
|
| 11 |
B |
3869 |
3646 |
25.15 |
|
|
|
| 12 |
B |
3774 |
3556 |
21.24 |
|
|
|
| 13 |
B |
1826 |
1721 |
44.07 |
|
|
|
| 14 |
B |
1428 |
1345 |
93.91 |
|
|
|
| 15 |
B |
1328 |
1251 |
0.04 |
|
|
|
| 16 |
B |
736 |
694 |
413.51 |
|
|
|
| 17 |
B |
506 |
477 |
35.42 |
|
|
|
| 18 |
B |
252 |
237 |
21.38 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 14888.8 cm
-1
Scaled (by 0.9422) Zero Point Vibrational Energy (zpe) 14028.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2=FULL/6-31G*
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.006 |
0.609 |
0.038 |
| C2 |
-0.006 |
-0.609 |
0.038 |
| N3 |
-0.006 |
1.969 |
-0.078 |
| N4 |
0.006 |
-1.969 |
-0.078 |
| H5 |
-0.324 |
2.459 |
0.750 |
| H6 |
0.854 |
2.369 |
-0.433 |
| H7 |
0.324 |
-2.459 |
0.750 |
| H8 |
-0.854 |
-2.369 |
-0.433 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
N4 |
H5 |
H6 |
H7 |
H8 |
| C1 | | 1.2188 | 1.3642 | 2.5805 | 2.0089 | 2.0089 | 3.1654 | 3.1355 |
C2 | 1.2188 | | 2.5805 | 1.3642 | 3.1654 | 3.1355 | 2.0089 | 2.0089 | N3 | 1.3642 | 2.5805 | | 3.9371 | 1.0133 | 1.0135 | 4.5160 | 4.4335 | N4 | 2.5805 | 1.3642 | 3.9371 | | 4.5160 | 4.4335 | 1.0133 | 1.0135 | H5 | 2.0089 | 3.1654 | 1.0133 | 4.5160 | | 1.6724 | 4.9597 | 4.9982 | H6 | 2.0089 | 3.1355 | 1.0135 | 4.4335 | 1.6724 | | 4.9982 | 5.0360 | H7 | 3.1654 | 2.0089 | 4.5160 | 1.0133 | 4.9597 | 4.9982 | | 1.6724 | H8 | 3.1355 | 2.0089 | 4.4335 | 1.0135 | 4.9982 | 5.0360 | 1.6724 | |
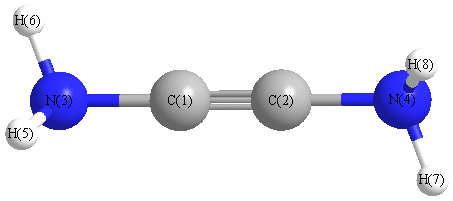 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
N4 |
175.408 |
|
C1 |
N3 |
H5 |
114.768 |
| C1 |
N3 |
H6 |
114.723 |
|
C2 |
C1 |
N3 |
175.408 |
| C2 |
N4 |
H7 |
114.768 |
|
C2 |
N4 |
H8 |
114.723 |
| H5 |
N3 |
H6 |
111.718 |
|
H7 |
N4 |
H8 |
111.718 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at G2
| | hartrees |
|---|
| Energy at 0K | -187.723546 |
| Energy at 298.15K | -187.717804 |
| HF Energy | -186.867258 |
| Nuclear repulsion energy | 103.045407 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/6-31G*
Geometric Data calculated at MP2=FULL/6-31G*
Point Group is C2
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability