Jump to
S1C2
S1C3
Energy calculated at MP3=FULL/cc-pV(T+d)Z
| | hartrees |
|---|
| Energy at 0K | -1591.278606 |
| Energy at 298.15K | |
| HF Energy | -1590.156321 |
| Nuclear repulsion energy | 352.085211 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP3=FULL/cc-pV(T+d)Z
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
538 |
538 |
0.00 |
|
|
|
| 2 |
A1 |
204 |
204 |
0.00 |
|
|
|
| 3 |
B1 |
494 |
494 |
0.00 |
|
|
|
| 4 |
B2 |
319 |
319 |
0.00 |
|
|
|
| 5 |
E |
464 |
464 |
0.04 |
|
|
|
| 5 |
E |
464 |
464 |
0.04 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 1241.1 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 1241.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP3=FULL/cc-pV(T+d)Z
Point Group is D2d
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.000 |
1.426 |
0.308 |
| S2 |
0.000 |
-1.426 |
0.308 |
| S3 |
-1.426 |
0.000 |
-0.308 |
| S4 |
1.426 |
0.000 |
-0.308 |
Atom - Atom Distances (Å)
| |
S1 |
S2 |
S3 |
S4 |
| S1 | | 2.8513 | 2.1080 | 2.1080 |
S2 | 2.8513 | | 2.1080 | 2.1080 | S3 | 2.1080 | 2.1080 | | 2.8513 | S4 | 2.1080 | 2.1080 | 2.8513 | |
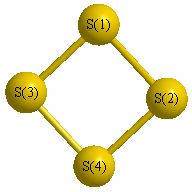 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| S1 |
S2 |
S4 |
47.444 |
|
S1 |
S3 |
S4 |
47.444 |
| S2 |
S1 |
S3 |
47.444 |
|
S2 |
S4 |
S3 |
47.444 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C3
Energy calculated at MP3=FULL/cc-pV(T+d)Z
| | hartrees |
|---|
| Energy at 0K | -1591.290618 |
| Energy at 298.15K | |
| HF Energy | -1590.133147 |
| Nuclear repulsion energy | 334.194101 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP3=FULL/cc-pV(T+d)Z
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
701 |
701 |
4.55 |
|
|
|
| 2 |
A1 |
431 |
431 |
5.52 |
|
|
|
| 3 |
A1 |
139 |
139 |
0.10 |
|
|
|
| 4 |
A2 |
212 |
212 |
0.00 |
|
|
|
| 5 |
B2 |
752 |
752 |
3.85 |
|
|
|
| 6 |
B2 |
346 |
346 |
2.14 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 1290.4 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 1290.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP3=FULL/cc-pV(T+d)Z
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.000 |
1.035 |
0.907 |
| S2 |
0.000 |
-1.035 |
0.907 |
| S3 |
0.000 |
1.612 |
-0.907 |
| S4 |
0.000 |
-1.612 |
-0.907 |
Atom - Atom Distances (Å)
| |
S1 |
S2 |
S3 |
S4 |
| S1 | | 2.0692 | 1.9042 | 3.2091 |
S2 | 2.0692 | | 3.2091 | 1.9042 | S3 | 1.9042 | 3.2091 | | 3.2245 | S4 | 3.2091 | 1.9042 | 3.2245 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| S1 |
S2 |
S4 |
107.659 |
|
S1 |
S3 |
S4 |
72.341 |
| S2 |
S1 |
S3 |
107.659 |
|
S2 |
S4 |
S3 |
72.341 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
Energy calculated at MP3=FULL/cc-pV(T+d)Z
| | hartrees |
|---|
| Energy at 0K | -1591.278164 |
| Energy at 298.15K | |
| HF Energy | -1590.097509 |
| Nuclear repulsion energy | 338.168747 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP3=FULL/cc-pV(T+d)Z
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
795 |
795 |
0.00 |
|
|
|
| 2 |
Ag |
303 |
303 |
0.00 |
|
|
|
| 3 |
Au |
252 |
252 |
0.00 |
|
|
|
| 4 |
B1u |
1008 |
1008 |
3777.12 |
|
|
|
| 5 |
B2u |
206i |
206i |
19.64 |
|
|
|
| 6 |
B3g |
352 |
352 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 1251.5 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 1251.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP3=FULL/cc-pV(T+d)Z
Point Group is D2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.000 |
0.937 |
1.261 |
| S2 |
0.000 |
0.937 |
-1.261 |
| S3 |
0.000 |
-0.937 |
1.261 |
| S4 |
0.000 |
-0.937 |
-1.261 |
Atom - Atom Distances (Å)
| |
S1 |
S2 |
S3 |
S4 |
| S1 | | 2.5219 | 1.8748 | 3.1424 |
S2 | 2.5219 | | 3.1424 | 1.8748 | S3 | 1.8748 | 3.1424 | | 2.5219 | S4 | 3.1424 | 1.8748 | 2.5219 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| S1 |
S2 |
S4 |
90.000 |
|
S1 |
S3 |
S4 |
90.000 |
| S2 |
S1 |
S3 |
90.000 |
|
S2 |
S4 |
S3 |
90.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability