Jump to
S1C2
Vibrational Frequencies calculated at MP3=FULL/cc-pVDZ
Geometric Data calculated at MP3=FULL/cc-pVDZ
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at MP3=FULL/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -344.335842 |
| Energy at 298.15K | -344.352982 |
| HF Energy | -343.072797 |
| Nuclear repulsion energy | 423.565312 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP3=FULL/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3132 |
2945 |
0.00 |
|
|
|
| 2 |
A1 |
3105 |
2919 |
0.00 |
|
|
|
| 3 |
A1 |
1514 |
1424 |
0.00 |
|
|
|
| 4 |
A1 |
1387 |
1304 |
0.00 |
|
|
|
| 5 |
A1 |
1274 |
1198 |
0.00 |
|
|
|
| 6 |
A1 |
1027 |
965 |
0.00 |
|
|
|
| 7 |
A1 |
1014 |
954 |
0.00 |
|
|
|
| 8 |
A1 |
837 |
787 |
0.00 |
|
|
|
| 9 |
A1 |
610 |
573 |
0.00 |
|
|
|
| 10 |
A1 |
32 |
30 |
0.00 |
|
|
|
| 11 |
A2 |
3152 |
2964 |
0.00 |
|
|
|
| 12 |
A2 |
3090 |
2906 |
106.91 |
|
|
|
| 13 |
A2 |
1500 |
1411 |
5.34 |
|
|
|
| 14 |
A2 |
1401 |
1318 |
7.70 |
|
|
|
| 15 |
A2 |
1205 |
1133 |
0.00 |
|
|
|
| 16 |
A2 |
1013 |
952 |
23.91 |
|
|
|
| 17 |
A2 |
806 |
758 |
0.00 |
|
|
|
| 18 |
A2 |
798 |
750 |
63.89 |
|
|
|
| 19 |
E |
3159 |
2970 |
81.38 |
|
|
|
| 19 |
E |
3159 |
2970 |
81.38 |
|
|
|
| 20 |
E |
3135 |
2948 |
0.00 |
|
|
|
| 20 |
E |
3135 |
2948 |
0.00 |
|
|
|
| 21 |
E |
3098 |
2913 |
101.93 |
|
|
|
| 21 |
E |
3098 |
2913 |
101.93 |
|
|
|
| 22 |
E |
3087 |
2903 |
0.00 |
|
|
|
| 22 |
E |
3087 |
2903 |
0.00 |
|
|
|
| 23 |
E |
1500 |
1410 |
7.81 |
|
|
|
| 23 |
E |
1500 |
1410 |
7.81 |
|
|
|
| 24 |
E |
1486 |
1397 |
0.00 |
|
|
|
| 24 |
E |
1486 |
1397 |
0.00 |
|
|
|
| 25 |
E |
1378 |
1296 |
13.09 |
|
|
|
| 25 |
E |
1378 |
1296 |
13.09 |
|
|
|
| 26 |
E |
1371 |
1289 |
0.00 |
|
|
|
| 26 |
E |
1371 |
1289 |
0.00 |
|
|
|
| 27 |
E |
1346 |
1266 |
0.20 |
|
|
|
| 27 |
E |
1346 |
1266 |
0.20 |
|
|
|
| 28 |
E |
1336 |
1256 |
0.00 |
|
|
|
| 28 |
E |
1336 |
1256 |
0.00 |
|
|
|
| 29 |
E |
1223 |
1150 |
0.00 |
|
|
|
| 29 |
E |
1223 |
1150 |
0.00 |
|
|
|
| 30 |
E |
1135 |
1068 |
35.43 |
|
|
|
| 30 |
E |
1135 |
1068 |
35.43 |
|
|
|
| 31 |
E |
1074 |
1010 |
0.00 |
|
|
|
| 31 |
E |
1074 |
1010 |
0.00 |
|
|
|
| 32 |
E |
930 |
875 |
6.31 |
|
|
|
| 32 |
E |
930 |
875 |
6.31 |
|
|
|
| 33 |
E |
837 |
787 |
2.92 |
|
|
|
| 33 |
E |
837 |
787 |
2.92 |
|
|
|
| 34 |
E |
595 |
560 |
0.00 |
|
|
|
| 34 |
E |
595 |
560 |
0.00 |
|
|
|
| 35 |
E |
428 |
402 |
0.02 |
|
|
|
| 35 |
E |
428 |
402 |
0.02 |
|
|
|
| 36 |
E |
329 |
309 |
0.00 |
|
|
|
| 36 |
E |
329 |
309 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 40893.0 cm
-1
Scaled (by 0.9403) Zero Point Vibrational Energy (zpe) 38451.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
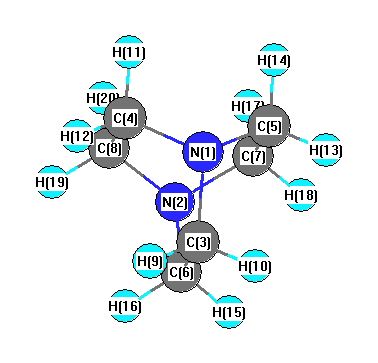
Geometric Data calculated at MP3=FULL/cc-pVDZ
Point Group is D3
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.000 |
1.297 |
| N2 |
0.000 |
0.000 |
-1.297 |
| C3 |
0.000 |
1.380 |
0.782 |
| C4 |
1.195 |
-0.690 |
0.782 |
| C5 |
-1.195 |
-0.690 |
0.782 |
| C6 |
-0.000 |
1.380 |
-0.782 |
| C7 |
-1.195 |
-0.690 |
-0.782 |
| C8 |
1.195 |
-0.690 |
-0.782 |
| H9 |
0.889 |
1.894 |
1.184 |
| H10 |
-0.889 |
1.894 |
1.184 |
| H11 |
1.196 |
-1.717 |
1.184 |
| H12 |
2.085 |
-0.178 |
1.184 |
| H13 |
-2.085 |
-0.177 |
1.184 |
| H14 |
-1.196 |
-1.717 |
1.184 |
| H15 |
-0.889 |
1.894 |
-1.184 |
| H16 |
0.889 |
1.894 |
-1.184 |
| H17 |
-1.196 |
-1.717 |
-1.184 |
| H18 |
-2.085 |
-0.178 |
-1.184 |
| H19 |
2.085 |
-0.177 |
-1.184 |
| H20 |
1.196 |
-1.717 |
-1.184 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
C3 |
C4 |
C5 |
C6 |
C7 |
C8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
| N1 | | 2.5945 | 1.4730 | 1.4730 | 1.4730 | 2.4952 | 2.4952 | 2.4952 | 2.0953 | 2.0953 | 2.0953 | 2.0953 | 2.0953 | 2.0953 | 3.2453 | 3.2454 | 3.2453 | 3.2454 | 3.2453 | 3.2454 |
N2 | 2.5945 | | 2.4952 | 2.4952 | 2.4952 | 1.4730 | 1.4730 | 1.4730 | 3.2453 | 3.2454 | 3.2453 | 3.2454 | 3.2453 | 3.2454 | 2.0953 | 2.0953 | 2.0953 | 2.0953 | 2.0953 | 2.0953 | C3 | 1.4730 | 2.4952 | | 2.3899 | 2.3899 | 1.5634 | 2.8559 | 2.8559 | 1.1026 | 1.1026 | 3.3437 | 2.6330 | 2.6330 | 3.3437 | 2.2174 | 2.2174 | 3.8576 | 3.2610 | 3.2609 | 3.8576 | C4 | 1.4730 | 2.4952 | 2.3899 | | 2.3899 | 2.8559 | 2.8559 | 1.5634 | 2.6330 | 3.3437 | 1.1026 | 1.1026 | 3.3437 | 2.6330 | 3.8576 | 3.2610 | 3.2610 | 3.8576 | 2.2174 | 2.2174 | C5 | 1.4730 | 2.4952 | 2.3899 | 2.3899 | | 2.8559 | 1.5634 | 2.8559 | 3.3437 | 2.6330 | 2.6330 | 3.3437 | 1.1026 | 1.1026 | 3.2610 | 3.8576 | 2.2174 | 2.2174 | 3.8576 | 3.2610 | C6 | 2.4952 | 1.4730 | 1.5634 | 2.8559 | 2.8559 | | 2.3899 | 2.3899 | 2.2174 | 2.2174 | 3.8576 | 3.2610 | 3.2609 | 3.8576 | 1.1026 | 1.1026 | 3.3437 | 2.6330 | 2.6330 | 3.3437 | C7 | 2.4952 | 1.4730 | 2.8559 | 2.8559 | 1.5634 | 2.3899 | | 2.3899 | 3.8576 | 3.2610 | 3.2610 | 3.8576 | 2.2174 | 2.2174 | 2.6330 | 3.3437 | 1.1026 | 1.1026 | 3.3437 | 2.6330 | C8 | 2.4952 | 1.4730 | 2.8559 | 1.5634 | 2.8559 | 2.3899 | 2.3899 | | 3.2610 | 3.8576 | 2.2174 | 2.2174 | 3.8576 | 3.2610 | 3.3437 | 2.6330 | 2.6330 | 3.3437 | 1.1026 | 1.1026 | H9 | 2.0953 | 3.2453 | 1.1026 | 2.6330 | 3.3437 | 2.2174 | 3.8576 | 3.2610 | | 1.7773 | 3.6239 | 2.3922 | 3.6239 | 4.1694 | 2.9602 | 2.3673 | 4.7946 | 4.3286 | 3.3655 | 4.3286 | H10 | 2.0953 | 3.2454 | 1.1026 | 3.3437 | 2.6330 | 2.2174 | 3.2610 | 3.8576 | 1.7773 | | 4.1694 | 3.6239 | 2.3922 | 3.6239 | 2.3673 | 2.9602 | 4.3286 | 3.3655 | 4.3286 | 4.7946 | H11 | 2.0953 | 3.2453 | 3.3437 | 1.1026 | 2.6330 | 3.8576 | 3.2610 | 2.2174 | 3.6239 | 4.1694 | | 1.7773 | 3.6239 | 2.3922 | 4.7946 | 4.3286 | 3.3655 | 4.3286 | 2.9602 | 2.3673 | H12 | 2.0953 | 3.2454 | 2.6330 | 1.1026 | 3.3437 | 3.2610 | 3.8576 | 2.2174 | 2.3922 | 3.6239 | 1.7773 | | 4.1694 | 3.6239 | 4.3286 | 3.3655 | 4.3286 | 4.7946 | 2.3673 | 2.9602 | H13 | 2.0953 | 3.2453 | 2.6330 | 3.3437 | 1.1026 | 3.2609 | 2.2174 | 3.8576 | 3.6239 | 2.3922 | 3.6239 | 4.1694 | | 1.7773 | 3.3655 | 4.3286 | 2.9602 | 2.3673 | 4.7946 | 4.3286 | H14 | 2.0953 | 3.2454 | 3.3437 | 2.6330 | 1.1026 | 3.8576 | 2.2174 | 3.2610 | 4.1694 | 3.6239 | 2.3922 | 3.6239 | 1.7773 | | 4.3286 | 4.7946 | 2.3673 | 2.9602 | 4.3286 | 3.3655 | H15 | 3.2453 | 2.0953 | 2.2174 | 3.8576 | 3.2610 | 1.1026 | 2.6330 | 3.3437 | 2.9602 | 2.3673 | 4.7946 | 4.3286 | 3.3655 | 4.3286 | | 1.7773 | 3.6239 | 2.3922 | 3.6239 | 4.1694 | H16 | 3.2454 | 2.0953 | 2.2174 | 3.2610 | 3.8576 | 1.1026 | 3.3437 | 2.6330 | 2.3673 | 2.9602 | 4.3286 | 3.3655 | 4.3286 | 4.7946 | 1.7773 | | 4.1694 | 3.6239 | 2.3922 | 3.6239 | H17 | 3.2453 | 2.0953 | 3.8576 | 3.2610 | 2.2174 | 3.3437 | 1.1026 | 2.6330 | 4.7946 | 4.3286 | 3.3655 | 4.3286 | 2.9602 | 2.3673 | 3.6239 | 4.1694 | | 1.7773 | 3.6239 | 2.3922 | H18 | 3.2454 | 2.0953 | 3.2610 | 3.8576 | 2.2174 | 2.6330 | 1.1026 | 3.3437 | 4.3286 | 3.3655 | 4.3286 | 4.7946 | 2.3673 | 2.9602 | 2.3922 | 3.6239 | 1.7773 | | 4.1694 | 3.6239 | H19 | 3.2453 | 2.0953 | 3.2609 | 2.2174 | 3.8576 | 2.6330 | 3.3437 | 1.1026 | 3.3655 | 4.3286 | 2.9602 | 2.3673 | 4.7946 | 4.3286 | 3.6239 | 2.3922 | 3.6239 | 4.1694 | | 1.7773 | H20 | 3.2454 | 2.0953 | 3.8576 | 2.2174 | 3.2610 | 3.3437 | 2.6330 | 1.1026 | 4.3286 | 4.7946 | 2.3673 | 2.9602 | 4.3286 | 3.3655 | 4.1694 | 3.6239 | 2.3922 | 3.6239 | 1.7773 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C3 |
C6 |
110.487 |
|
N1 |
C3 |
H9 |
108.022 |
| N1 |
C3 |
H10 |
108.022 |
|
N1 |
C4 |
C8 |
110.487 |
| N1 |
C4 |
H11 |
108.022 |
|
N1 |
C4 |
H12 |
108.022 |
| N1 |
C5 |
C7 |
110.487 |
|
N1 |
C5 |
H13 |
108.022 |
| N1 |
C5 |
H14 |
108.022 |
|
N2 |
C6 |
C3 |
110.487 |
| N2 |
C6 |
H15 |
108.022 |
|
N2 |
C6 |
H16 |
108.022 |
| N2 |
C7 |
C5 |
110.487 |
|
N2 |
C7 |
H17 |
108.022 |
| N2 |
C7 |
H18 |
108.022 |
|
N2 |
C8 |
C4 |
110.487 |
| N2 |
C8 |
H19 |
108.022 |
|
N2 |
C8 |
H20 |
108.022 |
| C3 |
N1 |
C4 |
108.437 |
|
C3 |
N1 |
C5 |
108.437 |
| C3 |
C6 |
H15 |
111.379 |
|
C3 |
C6 |
H16 |
111.380 |
| C4 |
N1 |
C5 |
108.437 |
|
C4 |
C8 |
H19 |
111.379 |
| C4 |
C8 |
H20 |
111.380 |
|
C5 |
C6 |
H15 |
101.560 |
| C5 |
C6 |
H16 |
151.027 |
|
C6 |
N2 |
C7 |
108.437 |
| C6 |
N2 |
C8 |
108.437 |
|
C6 |
C3 |
H9 |
111.379 |
| C6 |
C3 |
H10 |
111.380 |
|
C7 |
N2 |
C8 |
108.437 |
| C7 |
C5 |
H13 |
111.379 |
|
C7 |
C5 |
H14 |
111.380 |
| C8 |
C4 |
H11 |
111.379 |
|
C8 |
C4 |
H12 |
111.380 |
| H9 |
C3 |
H10 |
107.399 |
|
H11 |
C4 |
H12 |
107.399 |
| H13 |
C5 |
H14 |
107.399 |
|
H15 |
C6 |
H16 |
107.399 |
| H17 |
C7 |
H18 |
107.399 |
|
H19 |
C8 |
H20 |
107.399 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability